Indice
In chimica organica si sente parlare con estrema frequenza di elettrofilo e nucleofilo, oppure delle loro proprietà, l’elettrofilia e nucleofilia.
Si legge spesso, per fare un esempio, che una certa molecola è elettrofila, mentre un’altra è nucleofila, e magari quest’ultima è un nucleofilo più debole di un’altra specie chimica anch’essa nucleofila. Comprendiamo bene, già dalla ricorsività dei termini, che deve trattarsi di un importante concetto di base, al quale purtroppo la scuola secondaria di secondo grado non sempre e non sufficientemente è solita affrontare in modo adeguato. Alla base di questo vi è un’impostazione prevalentemente descrittiva della chimica organica, che punta ancora molto al raggiungimento di capacità nella nomenclatura delle molecole ed alla loro classificazione sistematica, cercando di leggere in questa chiave anche la reattività delle molecole organiche. La chimica delle sostanze organiche naturali e la biochimica, a loro volta, sembrano ritagliare sempre più quello spazio normalmente dedicato alla chimica organica, impoverendo ulteriormente i programmi dalla chimica organica di base. Ovviamente si tratta solo di osservazioni compiute su un numero limitato di scuole, in occasione dell’incontri didattici con singoli studenti, e la considerazione decade del tutto quando si parla di istituti tecnici a specifico indirizzo chimico.
Quelli di elettrofilo e nucleofilo sono concetti fondamentali al fine di:
– descrivere il meccanismo attraverso il quale si svolgono la maggior parte delle reazioni organiche (ma non esclusivamente), comprese ovviamente quelle in campo biochimico;
– poter individuare quali delle reazioni, tutte a loro modo “possibili sulla carta” possono dirsi possibili anche in chimica e quali no;
– descrivere le proprietà reattive di un gruppo funzionale o più in generale di una specie chimica;
– poter quantificare e prevedere l’entità della proprietà reattiva specifica di una molecola organica rispetto ad un’altra che manifesta la stessa tipologia di proprietà ma con “forza” differente.
Si tratta quindi di termini che comprendiamo fin da subito risultare fra loro complementari, che definiscono delle proprietà delle molecole, o meglio ancora quelli che in una chimica ritenuta più avanzata verrebbero definiti “descrittori molecolari”.
Proprietà che possono variare in modo continuo, da una condizione estrema per alcune specie chimiche ad una sostanzialmente neutrale per altre specie, fino ad arrivare ad una sorta di inversione nella proprietà, che a sua volta punta fino all’estremizzazione della proprietà nel senso contrario alla prima per altri tipi di molecole. Un caso che ci ricorda da vicino quello dei potenziali di ossidazione o di riduzione, o ancora più da vicino la scala per la misurazione del pH.
In effetti, sarà forse un caso, ma vi è un’attinenza assolutamente non con il pH bensì con i concetti di acido e di base introdotti da Lewis.
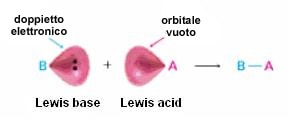
Al contrario della concezione di Brønsted-Lowry di acido come sostanza in grado di cedere e della base come sostanza in grado di acquistare ioni idrogeno (o idrogenioni, di fatto “protoni” H+), il chimico statunitense Gilbert Newton Lewis aveva optato per un’ulteriore modifica della definizione, tale da non puntare più l’attenzione sugli idrogenioni stessi, responsabili in qualche modo solo di una specie di “punta dell’iceberg” del ben più profondo effetto dell’acidità/basicità di una molecola, ma di valorizzare invece il ruolo degli elettroni di non-legame, i doppietti elettronici liberi che caratterizzano talune molecole, e sugli orbitali vuoti, ovvero non popolati da elettroni, che ne caratterizzano altre. Si definiscono basi di Lewis le specie chimiche con orbitali elettonici (una coppia) non impegnati in legame nel momento in cui donano questa coppia di elettroni ad un’altra specie chimica, detta base, contenente quest’ultima (detta base di Lewis) un orbitale vuoto, in grado di accettarli.
In chimica organica, il l’elettrofilo coincide di fatto con l’acido di Lewis, mentre il nucleofilo coincide di fatto con la base di Lewis.
La ragione per la quale si è scelto a ragione di non far banalmente coincidere i termini è relativa al diversa disciplina all’interno della chimica nel quale essi sono stati sviluppati: gli acidi e le basi di Lewis nello studio delle proprietà acido-base, appunto, che descrive lo stato di equilibrio di una reazione; i concetto di elettrofilo e nucleofilo descrittivo invece di una situazione in potenza, quella della “reattività” appunto, che è cosa completamente diversa dalla reazione già in parte avvenuta, ovvero in condizioni di equilibrio.
Un’altra ragione per la quale le due definizioni con sono equivalenti sta anche nel fatto che non tutte le specie chimiche organiche che si comportano come elettrofili o come nucleofili sono anche rispettivamente basi ed acidi di Lewis.
In chimica organica una molecola si comporta come elettrofilo quando è in grado di formare un legame chimico con il suo partner di reazione attraverso l’accettazione di una coppia di elettroni contenuti in orbitale atomico di quest’ultimo, detto nucleofilo.
ELETTROFILICITA’ E NUCLEOFILICITA’
Le due classi di specie chimiche che rappresentano la stragrande maggioranza degli ELETTROFILI sono costituite da molecole neutre con doppietti elettronici non condivisi e da cationi. In considerazione del fatto che possiamo avere
– Molecole organiche neutre nelle quali in carbonio si trovi in stato di carenza elettronica e quindi in grado di accettare doppietti elettronici, come ad esempio i composti carbonilici in genere (es. chetoni ed aldeidi), alogenuri acilici, alogenuri alchilici e talvolta anche alcheni ed alchini;
– Carbocationi organici (molecole con carica positiva);
– Molecole inorganiche neutre con doppietti elettronici non condivisi (come ad esempio il BH3);
– Molecole inorganiche neutre polarizzate (come CO2 ed HCl o polarizzabili come Br2);
– Cationi inorganici (ioni positivi, come ad esempio NO+ e H3O+).
Le due classi di specie chimiche che rappresentano la stragrande maggioranza dei NUCLEOFILI sono invece costituite da:
– Molecole organiche neutre con doppietti elettronici non condivisi (come ad esempio gli alcoli, gli eteri e le amine);
– Carboanioni organici (molecole con carica negativa);
– Molecole inorganiche neutre con doppietti elettronici non condivisi (come ad esempio l’H2O, NH3);
– Anioni inorganici (ioni carichi negativamente, come ad esempio OH–, Cl–, CO3-2).
A parità di classe chimica di appartenenza, specie chimiche diverse possono manifestare un più o meno marcato comportamento nucleofilo o elettrofilo in funzione della vicinanza di gruppi elettron-accettori, come ad esempio quelli ricchi di atomi elettronegativi, o elettron-donatori.
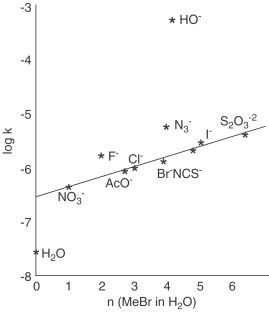
Numerosi gli approcci empirici proposti a partire dagli anni ’50 per poter quantificare almeno in senso relativo la forza della nucleofilicità o dell’elettrofilicità di una specie chimica. La maggior parte di queste relazioni (es. Swain-Scott, Ritchie, ecc) si basa sulla misura della velocità di reazione di attacco nucleofilo su di un substrato di riferimento (nello specifico della Swain-Scott il metil bromuro) rispetto ad un nucleofilo standard di riferimento, l’acqua. E’ possibile in questo modo estrapolare quanto una certa specie sia più o meno nucleofila di un’altra: ad esempio fatta 1 l’acqua, lo ione cloruro sarò 4.2, il benzil cloruro 0.87 ed il tiosolfato 6.4.
REAZIONI TRA NUCLEOFILO ED ELETTROFILO
L’interazione di tipo reattivo tra un elettrofilo ed un nucleofilo risulta di assoluta centralità nell’ambito della descrizione delle reazioni in chimica organica. Nella maggior parte dei casi, al di là del successivo decorso della reazione, il punto di partenza è infatti proprio la reazione tra un elettrofilo ed un nucleofilo, che porta alla formazione di nuove specie chimiche, per lo più instabili, soggette ad un’ulteriore evoluzione.
• Addizione nucleofila (su legami multipli carbonio-carbonio o carbonio-eteroatomo)
• Addizione elettrofila (di solito su legami multipli carbonio-carbonio)
• Sostituzione nucleofila (con molecolarità 1 o 2, alifatica o aromatica)
• Sostituzione elettrofila (per lo più aromatica)
Questo breve elenco non esaurisce la complessa casistica delle reazioni in chimica organica, pur occupandone una parte decisamente preponderante
Già intuitivamente, ovvero dal punto di vista terminologico, si comprende come le reazioni di “addizione” possano avvenire solo in condizioni di insaturazione del substrato, ovvero della molecola alla quale il reagente – nucleofilo o elettrofilo che sia – dovrà addizionarsi.
Potrà quindi trattarsi di reazioni a carico di legami multipli, doppi o tripli, come quelli tra due atomi di carbonio (alcheni ed alchini) o tra carbonio ed un eteroatomo, come ad esempio l’ossigeno (composti carbonilici come aldeidi e chetoni), azoto, zolfo, ecc.
Al contrario, le reazioni di “sostituzione” prevedono l’eliminazione di qualcosa, siano essi atomi, ioni o piccole molecole, dal substrato di reazione, con il contestuale inserimento (legame) dell’agente elettrofilo oppure nucleofilo al posto della porzione molecolare eliminata, detta gruppo uscente.

Risulta essere necessario ricordare come nella maggior parte dei casi un reagente elettrofilo o nucleofilo non può essere aggiunto “di per sé” al substrato, allo stesso modo con cui l’aggiunta di uno ione in chimica inorganica comportava anche il suo accompagnamento con un contro-ione di carica opposta. Nella maggior parte dei casi che seguono vedremo infatti come nelle reazioni di addizione, siano esse elettrofile oppure nucleofile, entrano in gioco seppur in momenti diversi l’agente principale che da il nome alla reazione, condizionandone l’andamento, insieme al suo necessario complemento. Può trattarsi di una coppia di ioni di carica opposta introdotti nel substrato come reagente unico (es. HCl) oppure di una molecola neutra, elettrofila o nucleofila, ma che per esplicare in pieno la sua azione richiede la co-presenza di un’altra specie molecolare di caratteristiche opposte.
Si ricorda infine che, dal punto di vista esteriore, ovvero dell’osservazione esterna del substrato e del suo prodotto, la classificazione che s’impone all’attenzione è quella sulla base fenomenologica, in questo caso addizione o sostituzione. Il fatto che dietro a questo epifenomeno vi sia un meccanismo di reazione che valorizza il ruolo di un nucleofilo piuttosto che di un elettrofilo potrebbe non emergere in una primissima istanza, qualora si osservasse solamente la molecola del substrato prima e dopo la sua reazione.
Al contrario, risulta chiaramente evidente già a prima vista se una reazione sia di addizione oppure di sostituzione. Una reazione di addizione, qualora si trascurino le specie transitorie o “incipienti”, si scrive nella sua globalità essenzialmente allo stesso modo sia che segua un meccanismo di addizione nucleofila oppure elettrofila:
Y-Z + -C=C- → Y-C-C-Z
Allo stesso modo una reazione di sostituzione, vista dal punto di vista della trasformazione del substrato, può essere comunque riportata come:
Y + -C-C-Z → -C-C-Y + Z
Addizione nucleofila
In questo caso si assiste allo smantellamento di un legame pigreco preesistente tra due atomi (di carbonio o tra un carbonio ed un eteroatomo) con la formazione da parte degli stessi due atomi di due nuovi legami sigma, dei quali uno nei confronti del nucleofilo.
Y-Z + -C=C- → Y-C-C-Z
Ad esempio, considerando l’addizione nucleofila su un gruppo carbonilico:
Si nota che la reazione avviene in due stadi: prima il nucleofilo attacca l’atomo di carbonio impoverito di elettroni e, legandosi ad esso, contribuisce alla formazione di un carbanione. Quest’ultimo, altamente reattivo, con la carica elettrica negativa formalizzata sul suo atomo più elettronegativo costituisce a sua volta un potente nucleofilo, tale da intercettare ogni molecola elettron-accettrice nei paraggi: nel caso riportato in esempio un idrogenione presente anch’esso nell’ambiente di reazione (es. derivante dallo stesso reagente introdotto inizialmente per fornire il nucleofilo Nu-).
Nel caso di legame carbonio-carbonio, perché il substrato risulti suscettibile all’attacco di un nucleofilo l’insaturazione deve trovarsi in una condizione di carenza di elettroni, ad esempio per la presenza vicinale di un gruppo fortemente elettron-attrattore, come ad esempio un alogeno.
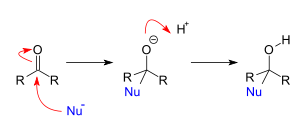
Nella pratica della sintesi di laboratorio, l’addizione nucleofila può risultare in competizione con l’addizione elettrofila dal momento che la specie che reagisce con il carbanione nella seconda fase di reazione è pur sempre un elettrofilo presente nell’ambiente di reazione. L’impiego di un mezzo solvente polare ma privo di idrogenioni scambiabili, ovvero di un solvente polare aprotico come il dimetilsolfossido o l’acetonitrile, può tornare di utilità nell’orientare in direzione nucleofila la reazione di addizione, dal momento che contribuisce a solvatare (e quindi a schermare) l’elettrofilo come ad esempio l’idrogenione dell’esempio precedente.
Addizione elettrofila
Anche in questo caso la reazione avviene a carico di un’insaturazione del substrato, che può consistere in un legame multiplo carbonio-carbonio oppure in carbonio-eteroatomo.
La reazione avviene formalmente in due stadi: il primo comporta il trasferimento della coppia di elettroni che inizialmente andavano a costituire il legame pigreco nel doppio legame del substrato, per andare a formare un legame semplice di tipo sigma con un reagente elettrofilo (nell’esempio Y+). Questo allontanamento di una coppia di elettroni porta la molecola inizialmente insatura ad acquisire una carica positiva, divenendo così un carbocatione con la carica positiva formalizzata sul un atomo di carbonio: è questa in effetti la vera specie chimica reattiva (elettrofilo) sulla quale si giocherà il successivo decorso della reazione.
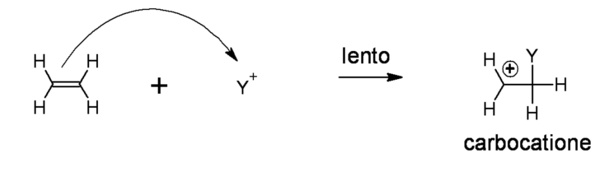
Nel decorso che segue è riportato il caso dell’addizione: l’elettrofilo di tipo carbocationico appena formato addiziona su di esso un nuclelofilo.
A fronte di catene più lunghe e complesse non è affatto escluso che il carbocatione evolva verso sistemi sempre carichi ma a maggiore stabilità, tramite la migrazione della carica elettrica laddove essa può essere meglio stabilizzata, ad esempio su altri sistemi aromatici o con elettroni pigreco o ancora su atomi di carbonio secondari. In questo caso il nucleofilo si legherà comunque all’atomo di carbonio sul quale in quel momento si viene a trovare la carica positiva, con la possibile formazione di una miscela di prodotti fra loro isomeri.
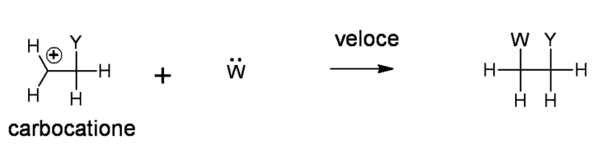
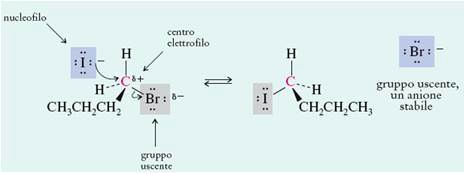
Sostituzione nucleofila
Questo caso ci ricorda da vicino come eravamo abituati a ragionare nella chimica inorganica nelle reazioni acido/base: un gruppo “più forte” (in questo caso nella sua nucleofilicità) scalza e quindi sostituisce da un certo substrato molecolare un gruppo più debole per la stessa proprietà.
![]()
In questo caso la cosa può anche essere vista in senso inverso: una reazione di sostituzione nucleofila risulta tanto più favorita quanto quello che viene eliminato può essere considerato “un buon gruppo uscente”.
La sostituzione nucleofila può a sua volta avvenire seguendo due distinti meccanismi, uno di tipo monomolecolare a due stadi (detto SN1), oppure l’altro bimolecolare ad un solo stadio concertato (detto Sn2).
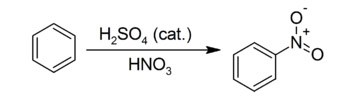
Sostituzione elettrofila
E’ questo il caso in cui un elettrofilo interagisce con il substrato portando all’espulsione da esso di altro gruppo, o anche di un singolo atomo, con il contestuale legame al posto di esso dello stesso elettrofilo.
Anche da questo tipo di reazioni vi è un “sottoprodotto”, ovvero una molecola uscente, che nell’esempio della nitrazione sopra riportata è H2O, formata dall’idrogenione espulso dal substrato aromatico insieme all’OH- “avanzato” dalla formazione dell’elettrofilo ione nitronio NO2+ a partire dall’acido nitrico.
Si tratta probabilmente della meno diffusa tra le tipologia di reazione appena riportate, caratterizzando in particolare la reattività (in qualità di substrati) delle molecole aromatiche, dove il gruppo uscente può essere costituito semplicemente da un atomo di idrogeno, o da particolari legami carbonio-carbonio insaturi come ad esempio i gruppi vinilici.
Attraverso le reazioni di addizione elettrofila è possibile superare l’ostacolo della grande stabilità (e quindi la scarsa reattività) dei sistemi aromatici come il benzene, introducendo in essi nuovi gruppi funzionali.