La cromatografia è fondamentalmente una tecnica che consente di separare sostanze chimiche fra loro diverse inizialmente in miscela, o meglio in soluzione, restituendole singolarmente nel tempo e, come implicito nel concetto stesso di “separazione” isolandole anche nello spazio.
Inutile dire che dietro a questa semplice definizione, che più generica davvero non si può, si nascondono decine di tecniche fra loro estremamente diverse in relazione ai principi chimici e chimico-fisici sfruttati, alle caratteristiche delle sostanze da separare e dei coadiuvanti chimici coinvolti, delle finalità della separazione e così via. Un piccolo mondo interno alla chimica che attraversa il primo luogo il campo dell’analisi per sfociare in applicazioni preparative sia su scala da laboratorio che industriale e che coinvolge una percentuale insospettabilmente alta degli addetti chimici di un po’ tutti i settori produttivi e di controllo: dal campo medico a quello agronomico, da quello ambientale a quello petrolchimico, da quello farmaceutico a quello ambientale e così via.
Tornando alla definizione inizialmente fornita, la cromatografia non è in effetti l’unico approccio possibile per realizzare una separazione come quella descritta. L’elettroforesi per esempio potrebbe rispondere anch’essa molto bene a questa definizione, e persino la distillazione e la cristallizzazione frazionata se opportunamente gestite possono essere utilizzate per la separazione di sostanze chimiche diverse in miscela, o meglio in soluzione, restituendole nel tempo in forma virtualmente pura, ovvero separandole l’una dall’altra.
Quello che caratterizza il complesso delle tecniche cromatografiche è però relativo alla modalità secondo la quale questa separazione è effettuata. Tutte le tecniche cromatografiche si basano sulla capacità delle singole specie chimiche contenute nella miscela di ripartirsi in modo differente tra una fase detta “stazionaria”, ovvero una sostanza chimica che possiamo considerare virtualmente immobile, ed un media di trasporto differente, costituito da un’altra sostanza chimica che si muove rispetto alla prima ed è pertanto detta “fase mobile”.
Tutto qui. Una sostanza chimica in grande quantità, allo stato solido oppure liquido ma in qualche modo ferma, legata, stazionaria appunto, ed una quantità altrettanto grande di un’altra sostanza chimica, quest’ultima necessariamente fluida (liquida o gassosa) che vi passa sopra, o vi scorre attraverso. Le sostanze da separare, introdotte invece sempre in piccola quantità, devono potersi sciogliere completamente nella fase mobile e devono inoltre avere la capacità di interagire in qualche modo, secondo un qualsiasi principio chimico o chimico-fisico, con la fase stazionaria.
Se immaginiamo di organizzare tutto questo come un tubo, possiamo riempire questo tubo con dei granelli regolari, magari delle minuscole sferette, di un materiale solido (la fase stazionaria) e far passare attraverso di esso un liquido (la fase mobile), per caduta o pompandolo attivamente. Introducendo la miscela di sostanze da separare nel punto di ingresso della fase mobile, se un componente chimico della miscela non mostra alcun tipo di interazione con la fase stazionaria uscirà alla stessa velocità della fase mobile nella quale invece si scioglie molto bene; al contrario, se dovesse interagire in modo fortissimo, praticamente irreversibile, con la fase stazionaria si fisserebbe irrimediabilmente su questa e non uscirebbe più dal tubo.
In un sistema cromatografico ben organizzato, si scelgono le condizioni di lavoro (essenzialmente la natura chimica delle fasi stazionaria e mobile, ed eventualmente la velocità di quest’ultima, il pH, la temperatura e poco altro) per far sì che le sostanze da separare interagiscano sì, ma in modo reversibile con la fase stazionaria. Nel corso del tempo, man mano che la fase mobile fluisce, si assisterà pertanto ad un passaggio continuo e ripetuto delle singole sostanze della miscela dalla fase mobile a quella stazionaria e viceversa: più una sostanza tenderà ad interagire con la fase stazionaria più passerà del tempo legata a questa e tarderà la sua uscita dal tubo insieme alla fase mobile, che nel frattempo continua a fluire regolarmente.
Quello che ne risulterà sul lungo periodo, magari nel corso dei minuti, sarà un ritardo differente nell’uscita dal tubo delle sostanze che componevano la miscela introdotta, con il risultato di una separazione nel tempo delle stesse sostanze in soluzione. L’uscita di ogni singola sostanza non sarà concentrata in un istante infinitesimo ma occuperò un piccolo lasso di tempo, preferibilmente sull’ordine di grandezza dei secondi, lasso di tempo entro il quale la concentrazione della stessa sostanza salirà velocemente fino ad un massimo, per poi scemare in modo altrettanto veloce, descrivendo rispetto alla coordinata del tempo una curva di concentrazione simile ad una gaussiana.
Il tempo impiegato da ciascun componente della miscela impiegato per percorrere l’intero sistema, dal momento della sua introduzione a quello della sua uscita, valutato sul massimo di questa gaussiana, prende il nome di tempo di ritenzione e, a parità di tutto l’allestimento cromatografico e dei metodi utilizzati (che saranno di seguito descritti più nel dettaglio) dipende dalla natura della specie chimica in questione, quindi dalla sua struttura molecolare.
Se decidessimo di raccogliere “man mano” la fase mobile (in questo caso un liquido) in uscita dal sistema, ad esempio preparando una serie di provette allineate e passando a riempire una provetta dopo l’altra, otterremo una composizione diversa per ciascuna provetta, o come le definirebbero gli addetti alla cromatografia, per ogni “frazione” raccolta. In effetti proprio di frazioni si tratta, in quanto la miscela delle diverse sostanze, a volte poche, in altri casi anche centinaia, introdotte in testa al sistema, viene in questo modo frazionata, consentendo nel caso ideale di isolare le singole sostanze componenti, o più semplicemente in tanti altri casi di raccogliere frazioni un po’ meno complesse della miscela iniziale, frazioni magari composte giusto da un paio di componenti che eluiscono insieme, che rispetto alla condizione iniziale è comunque una grande semplificazione.
Eluiscono? Cosa significa questo termine? Si tratta forse di un errore di stampa? No, il verbo “eluire” è in effetti uno dei termini più utilizzati dai tecnici cromatografici, e significa semplicemente “uscire dal sistema cromatografico insieme alla fase mobile”; la stessa fase mobile è detta eluente, e tutto il processo di uscita dei singoli componenti, sciolti all’interno della fase mobile, è detto anche eluizione.
Sempre volendo conoscere meglio le peculiarità ricche di aneddoti del linguaggio della cromatografia, quello che finora abbiamo semplicemente definito come “tubo” prende in effetti il nome di colonna. La ragione riede nel fatto che le prime cromatografie realizzate alcune decine di anni fa erano realizzate all’interno di grandi contenitori cilindrici in vetro, molto alti rispetto al loro diametro, che avevano per l’appunto l’aspetto di piccole colonne e che venivano riempiti con una fase stazionaria composta da granelli di silice o di altro materiale come ad esempio saccarosio o talco. Anche se in seguito sono stati realizzati contenitori cromatografici di tutte le fogge e dimensioni, dai tubicini di pochi centimetri e di tutti i tipi di materiali a cilindri tozzi dove l’altrezza non supera poi di molto il diametro, o addirittura in matasse di un tubicino capillare sottilissimo che a vederlo assomiglia di più che altro ad un filo di rame smaltato, il termine “colonna” è pur sempre rimasto, per cui ancora oggi la colonna, qualunque essa sia, è e resta il luogo dove la separazione cromatografica si svolge. Pur all’interno di un’apparecchiatura più grande, costosa e complessa, ricca di controlli, automatismi e regolazioni, ai nostri giorni immancabilmente interfacciata un computer, è sempre nella colonna che si realizza la separazione cromatografica fra le sostanze inizialmente introdotte.
L’atto stesso dell’introduzione dev’essere unico, rapido e deciso: una piccola dose di miscela da separare, meglio se opportunamente sciolta in una quantità opportuna di solvente, magari della stessa fase mobile. Sarà anche per il fatto che, almeno sulle piccole attrezzature di laboratorio con finalità analitiche, questa introduzione la si realizza per mezzo di una siringa, ma questo atto prende immancabilmente il nome di “iniezione”.
Infine, il termine stesso “cromatografia”, o meglio la sua etimologia in un certo qual modo fuorviante, frutto del contesto storico e a dire il vero un po’ episodico che avvolge la sua scoperta. Letteralmente “cromatografia” significherebbe “scrittura con il colore”, ma cosa c’entrano, vi chiederete voi a questo punto, sia il colore che il concetto stesso di scrittura con quanto è stato descritto finora? Tutto dipende, come si accennava, da come l’effetto cromatografico è stato scoperto (dal chimico Michail Cvet, di origini italo-russo, nel 1906) e come questo è stato applicato almeno nei primi anni.
Immaginate che le diverse sostanze chimiche che, sciolte tutte insieme in un solvente, costituiscono la nostra miscela di partenza, siano sostanze intensamente colorate, magari ognuna di un colore differente: potete ottenere facilmente una soluzione del genere per esempio estraendo dei fiori o dei frutti colorati, o anche le foglie di una pianta verde. E’ facile intuire che le varie frazioni raccolte in uscita dalla colonna avranno colorazione fra loro differente, che rispecchierà la composizione delle frazioni isolate: la frazione dove sarà stato isolato il beta-carotene sarà colorata in giallo-arancio, quella con la clorofilla A sarà verde, quella dove è stata raccolta un’antocianidina magari sarà rosa e le frazioni nelle quali la fase mobile stava uscendo senza aver eluito alcun componente saranno semplicemente incolori. Ed ecco spiegata la ragione del riferimento al colore: il chimico Cvet lavorò proprio su un estratto di origine vegetale, dal quale riuscì ad isolare la clorofilla in forma pura. Per realizzare questa separazione utilizzò una colonna in vetro (quindi trasparente) riempita da una fase stazionaria costituita da minuscoli granuli di argilla, e come fase mobile una sostanza altamente apolare, l’etere di petrolio, che scioglie bene tutti i pigmenti estratti dalla pianta e non intacca l’argilla stessa. Dopo aver introdotto il campione di estratto nella parte sommitale (come si sul dire “in testa”) della colonna, iniziò ad aggiungere gradualmente etere di petrolio, notando così, grazie alla trasparenza del vetro, come man mano che questo liquido scendeva attraverso l’argilla, il campione di estratto vegetale scendeva si muoveva anch’esso dall’alto al basso all’interno della colonna, seppur con un certo ritardo rispetto all’etere di petrolio. In particolare notò che nel corso del tempo esso si differenziava in bande, ovvero in strati colorati differenti che, scendendo a velocità differente, si separavano sempre di più nella colonna. Da questa sorta di disegno costituito da strati di colore separati e disposti l’uno sotto l’altro, prese inizialmente origine il riferimento alla scrittura contenuto nella parola “cromatografia”.
![]()
MECCANISMI DI INTERAZIONE E FASI

Più che una tecnica specifica, la cromatografia può essere considerata un approccio metodologico alla separazione delle miscele di sostanze chimiche in soluzione. Esistono corsi specifici all’interno dei programmi di chimica, centinaia di libri in tutte le lingue e decine di riviste scientifiche internazionali, decine di ditte specializzate nella produzione e commercializzazione di apparecchiature, colonne e consumabili di vario tipo, nonché associazioni scientifiche e simposi annuali specificamente dedicati all’argomento ed alle ultime novità del settore. L’esistenza stessa di un ricco vocabolario di termini nati o adattati nel contesto cromatografico, denota già di per sé la ricchezza di questo campo, che da speculazione nata nell’ambito della tecnologia delle separazioni è ora diventato a tutti gli effetti uno degli argomenti più ampiamente affrontati da una consistente fetta di chimici nella loro attività quotidiana.
Un primo grossolano criterio di classificazione dei metodi cromatografici si basa sullo stato fisico della fase mobile utilizzata: quando la fase mobile è un liquido si parla di cromatografia liquida (LC), mentre quando essa è gassosa (solitamente costituita da elio, oppure idrogeno, o più raramente azoto) la tecnica prende il nome di gascromatografia (GC).
Sono state proposte e da taluni impiegate anche tecniche cromatografiche che utilizzano in qualità di fase mobile un fluido in condizioni supercritiche.
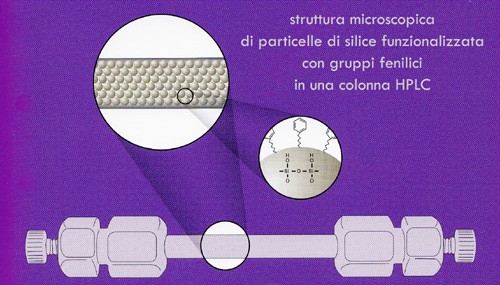
Nella cromatografia liquida la fase stazionaria (o fissa) è necessariamente un solido, nei più dei casi un materiale costituito da particelle sferiche di diametro piccolissimo (da 1.5 ad un centinaio di micron in media), solitamente regolari e molto ben impaccate per ridurre il più possibile la presenza di spazi morti fra le particelle. Fra i materiali più comunemente utilizzati per la loro costruzione figurano la silice ed i polimeri a base stirenica (polistirene/divinilbenzene), solitamente dotati di una porosità controllata all’interno di ciascun granulo (diametro dei pori da 120 a 200 angstrom circa).
Questi materiali possono essere in grado di stabilire interazioni con i soluti da sottoporre a separazione in modo diretto, o meglio ancora attraverso uno strato funzionalizzato, ovvero una struttura molecolare differente che il produttore stesso della resina provvede a legare chimicamente tramite opportune reazioni ai granuli di silice o di resina stirenica. Fra le funzionalizzazioni più comunemente utilizzate si ricordano la cosiddetta “C18” costituita dalla catena lineare satura dell’alacano ottadecano, oppure catene alchiliche terminanti con un gruppo aminico, o anche radicali fenilici legati a brevi catene alchiliche, e così via.
Esistono però anche fasi stazionarie non particellari, bensì estruse, che assumono la forma di cilindretti porosi contenenti al loro interno finissimi percorsi tortuosi; fasi stazionarie “depositate” su superfici rigide ed inerti, come si realizza per esempio nelle lastrine utilizzate nell’applicazione detta TLC (Thin Layer Chromatography) e addirittura un pezzo di carta porosa può essere utilizzato come fase stazionaria, in funzione della cellulosa che la compone, per effettuare su di esso una separazione cromatografica.
Nel campo della gascromatografia la fase stazionaria può invece essere sia di tipo solido (in questo caso anch’essa particellare) che, oggi soluzione ben più comune e performante, di tipo liquido.
Il lavorare con una fase stazionaria liquida comporta tuttavia due ordini di problemi: in primo luogo essa non deve essere asportata con il flusso del gas in transito (fase mobile), neppure ad alta temperatura. Per questo oltre a trattarsi di un liquido davvero alto bollente, esso viene chimicamente legato al materiale inerte del quale è costituita la colonna, solitamente in vetro. In secondo luogo, non potendo essere poroso, un liquido interagisce con il materiale in transito sulla sua superficie solo in funzione dell’estensione della superficie medesima. Per massimizzare questa interazione la forma delle colonne per cromatografia gas-liquido è andata nel tempo estremizzandosi, accrescendosi sempre più in lunghezza ed assottigliandosi in diametro e spessore dello strato di liquido legato, fino ad assumere la forma di un sottile tubicino (es. diametro interno 0.25 mm) “spalmato” al suo interno di un sottilissimo strato di fase stazionaria liquida legata (es. 0.25 micron) e lungo fino a 60 metri. Inutile dire che non disponendo ogni laboratorio di una struttura simile al CERN di Ginevra, questo tubicino, piuttosto flessibile per quanto costitito esternamente da vetro, è fornito già in forma arrotolata ed appoggiato su gancetti, così da assomigliare ad una piccola matassa di filo elettrico smaltato.
Sulla composizione chimica delle fasi stazionarie utilizzate in gascromatografia i materiali che la fanno da padroni sono derivati organici del silicio, i cosiddetti silos sani, come ad esempio il polimetildisilossano (PDMS) ed i suoi derivati che vedono una certa percentuale dei residui metilici di questa molecola sostituiti con altrettanti residui fenilici.
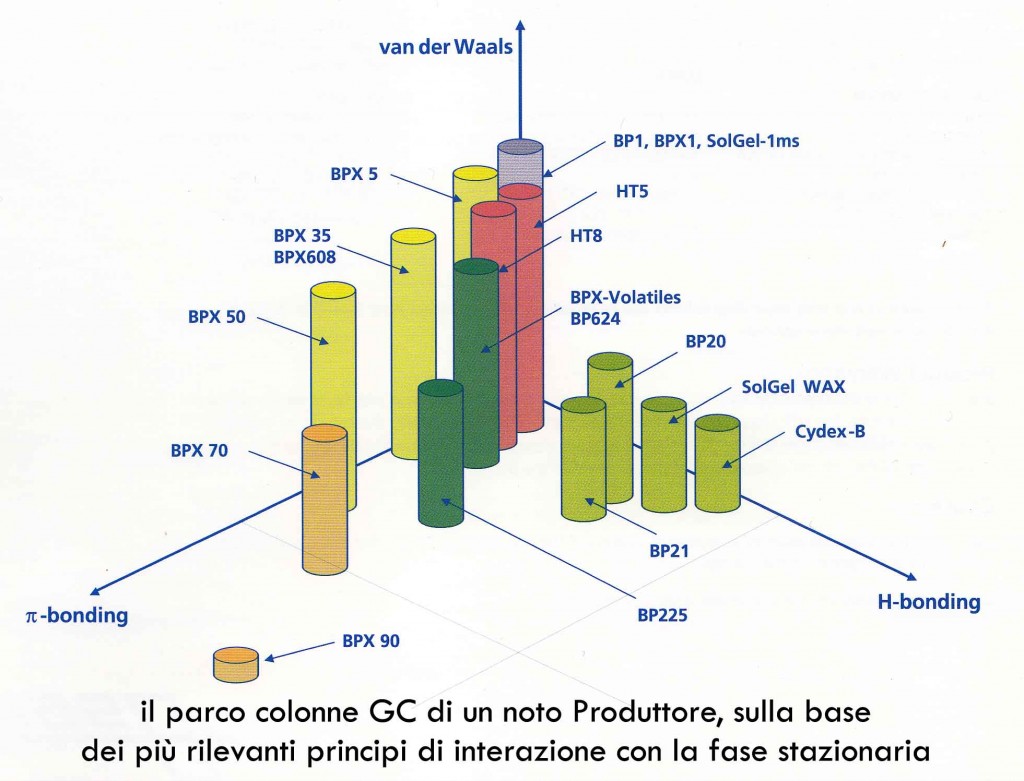
Per quanto riguarda i meccanismi di interazione tra le molecole da separare e le fasi mobile e stazionaria, possono essere sfruttati un po’ tutti i meccanismi di natura chimica e chimico-fisica dei quali siamo a conoscenza. La differenza di interazione fra le diverse molecole che compongono la miscela si realizza per lo più nei confronti della fase stazionaria, mentre la fase mobile rappresenta di solito un mezzo di trasporto passivo, nel quale le stesse molecole si sciolgono agevolmente e concorrenzialmente rispetto alla stessa fase stazionaria e che, essendo in movimento rispetto a quest’ultima, consente il deflusso delle stesse sostanza al di fuori della colonna. Nel caso della gascromatografia, addirittura, la fase stazionaria è spesso costituita da un gas inerte come l’elio: in un sistema del genere, anche grazie alle altissime temperature utilizzate (spesso >350°C) le molecole da separare si muovono nella fase mobile sotto forma di vapore, e sotto forma di vapore entrano in contatto con la fase stazionaria liquida legata, sciogliendosi in essa.
Tra i meccanismi di interazione possiamo avere una interazione polare, tramite la realizzazione di legami idrogeno, una repulsione idrofobica, interazioni di Van der Waals, interazioni pi-greco/pi-greco tra anelli aromatici presenti sulla fase stazionaria con analoghi anelli aromatici contenuti nelle nostra sostanze, scambio ionico (anionico e cationico), chelazione, esclusione dimensionale basata su proprietà steriche (in primo luogo sul diametro medio della molecola in soluzione), ecc.
In ogni caso quello che verrà a realizzarsi sarà una ripartizione tra la fase mobile e la fase stazionaria, caratterizzata da una costante di ripartizione (K) del tutto analoga a quella che si riscontra nella ripartizione di una specie chimica tra due fasi liquide fra loro immiscibili. In condizioni di equilibrio, questa costante non dipende dalla quantità di partenza della sostanza o da quanto essa era inizialmente abbondante in una delle due fasi, ma unicamente dalla natura chimica della terna costituita da fase mobile, fase stazionaria e molecola da separare. Una volta fissate le prime due variabili, ovvero scelto il sistema cromatografico di riferimento, la costante di ripartizione sarà funzione unicamente della natura chimica della molecola da separare. In una miscela costituita anche da molti componenti, ciascuna specie chimica dovrebbe avere almeno in linea di principio una costante di ripartizione differente, che si ripercuoterà sul suo diverso fattore di ritardo nell’uscita dalla colonna.
In effetti, almeno dal punto di vista didattico, lo studio degli equilibri di ripartizione di una sostanza tra due fasi immiscibili risulta propedeutico alla comprensione dei meccanismi che possono stare alla base della cromatografia. Anche i meccanismi di ritenzione differenziale fra le diverse molecole che non possono in effetti essere assimilati con una ripartizione tra fasi differenti, possono comunque essere perfettamente descritti in relazione ad un qualche tipo di costante di equilibrio (es. costante di formazione di dissociazione di una coppia ionica, costante di formazione di un complesso, ecc) numericamente differente per ciascun componente della miscela da separare.
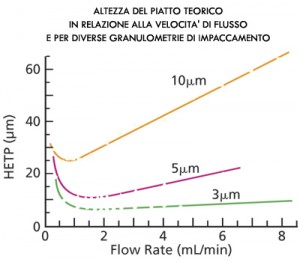
Propedeutica alla comprensione più profonda della cromatografia è tradizionalmente anche la teoria della distillazione, quella che fa riferimento alle colonne utilizzate in ambito industriale per la distillazione, costituite da piatti sovrapposti ove si realizzano singoli e successivi equilibri tra fase vapore e fase liquida. In questo caso la separazione fra le sostanze in miscela è basata com’è facilmente intuibile su un diverso punto di ebollizione (o tensione di vapore) ma per rendere la distillazione frazionata davvero efficiente è necessario che si realizzino una serie di equilibri fra i due stati fisici della stessa sostanza, in settori della colonna a temperatura differente, corrispondenti ai cosiddetti piatti di distillazione. Da qui il concetto di “piatto teorico” tradizionalmente utilizzato per quantificare comparativamente l’efficienza separativa anche delle colonne cromatografiche: anche se in cromatografia una colonna è un continuo e non è di certo divisa in settori, tantomeno in piatti, possiamo comunque immaginare che lo sia, sulla base delle performances separative ottenibili su miscele standardizzate di sostanze. Un colonna con un maggior numero di piatti teorici sarà maggiormente performante, in quanto riuscirà a separare meglio sostanze anche molto simili tra loro, per struttura molecolare e quindi per proprietà chimico-fisiche, rispetto ad una con un numero minore di piatti teorici. Allo stesso modo, a parità di lunghezza complessiva della colonna cromatografica, sarà obiettivo del costruttore pervenire alla massima riduzione possibile dell’altezza del piatto teorico, in modo tale da rendere maggiormente performante la separazione nell’unità di spazio.
Dal punto di vista della definizione, l’altezza del piatto teorico è la costante di proporzionalità tra la varianza della banda (si ricordi l’approssimazione gaussiana del profilo di concentrazione della sostanza in corso di separazione, rispetto al tempo) e la distanza che essa ha percorso all’interno della colonna.
L’altezza del piatto teorico (HETP), espressa in micron, è bene espressa dall’equazione di Van Deemter in relazione a diversi fattori, in primo luogo alla velocità di flusso della fase mobile ed al diametro medio dei granuli dell’impaccamento della fase stazionaria.
Anche se l’esame delle singole tecniche di separazione cromatografica, basate su principi di interazione chimico-fisici anche molto differenti fra loro, esula dagli obiettivi di questo articolo, vogliamo comunque fornire un breve elenco dei più utilizzati principi di separazione cromatografica, per consentire al lettore di effettuare gli opportuni approfondimenti ove riterrà necessario. Questi principi di separazione si riflettono in altrettante declinazioni tecnologiche dell’approccio alla separazione cromatografica.
CROMATOGRAFIA LIQUIDA E GASCROMATOGRAFIA
La distinzione principale fra le tecniche cromatografiche di basa sullo stato fisico della fase mobile: liquida, gassosa o, più raramente, supercritica.
La CROMATOGRAFIA LIQUIDA è solitamente intesa come la ripartizione delle molecole facenti parte della miscela iniziale in soluzione, fra una fase stazionaria di tipo solido ed una fase mobile di tipo liquido, anche se la fase stazionaria “attiva” può essere costituita da un liquido chimicamente legato ad un supporto solido (come nelle usatissime colonne con fase ottadecilica legata a granuli di silice).
A seconda della maggiore o minore polarità reciproca fra fase mobile e stazionaria possiamo distinguere l’approccio cromatografico-liquido in “fase diretta” (quando una fase mobile apolare, es. composta da un idrocarburo, si muove attraverso una fase stazionaria più polare, ad es. composta da silice) ed in “fase inversa” (quando una fase mobile più polare, es. metanolo, aceto nitrile, acqua si muove attraverso una fase stazionaria meno polare, es. un idrocarburo alifatico o aromatico legato ad un supporto solido). La cromatografia liquida in fase diretta è detta così semplicemente perché è stata la prima ad essere sviluppata storicamente, facendo uso di materiali già ampiamente disponibili già all’inizio del secolo.
Possiamo inoltre osservare applicazioni diverse della cromatografia liquida, diversificate per scopi e per disponibilità di mezzi tecnologici: dalla tradizionale cromatografia liquida (LC) per semplice percolazione gravitazionale della fase mobile, o sulla spinta di una modesto sistema di pompaggio a bassa pressione avente finalità essenzialmente di sgrossatura nella separazione di miscele complesse o di purificazione di prodotti da semplici contaminanti, si passa ad applicazioni ad alto contenuto tecnologico, tipiche del settore analitico, che sfruttano pressioni fino ad 400 bar pur lavorando su modestissime quantità di campione, l’HPLC (High Pressure/Performances Liquid Chromatography), o ancora più elevati nelle moderne tecniche UPLC.
Sulla base degli specifici meccanismi di interazione nell’ambito della terna costituita da sostanze da separare/fase stazionaria/fase mobile possiamo incisivamente riconoscere l’esistenza delle seguenti tecniche di separazione cromatografica in fase liquida:
Formazione di coppie ioniche
Ripartizione (a fase legata e non legata)
Esclusione
Ionica (con o senza soppressione)
Affinità
Fasi chirali
Scambio di legante
Una volta definita la finalità, l’assetto strumentale, il principio di separazione (vedi elenco appena sopra), l’eventuale condizione di lavoro in fase diretta/inversa, la fase stazionaria e la fase mobile, si può scegliere se lavorare nel modo cosiddetto “isocratico” (letteralmente “alla stessa concentrazione”), dove le condizioni operative non cambiano nel corso dell’eluizione, o “in gradiente” dove uno o più parametri di processo variano in modo graduale o discontinuo.
Tra i parametri più frequentemente oggetto di variazione nel corso dell’eluzizione figurano al primo posto la composizione della fase mobile: si può passare da una fase mobile costituita da un solvente A ad uno costituita da un solvente B in modo graduale, con i rapporti relativi tra i due solventi che cambia gradatamente nel tempo, oppure “a scalini” o scaglioni di concentrazione, tramite distemi di dosaggio volumetrico estremamente sofisticati, specie quando devono lavorare nelle condizioni di elevata pressione tipici dell’HPLC.
Altri parametri soggetti a variazione nel corso dell’eluizione sono la velocità del flusso della fase mobile e più raramente la temperatura, che ha per lo più la funzione di fluidificare maggiormente la fase mobile, riducendone la resistenza al passaggio attraverso la fase stazionaria qualora questa risultasse costituita da granuli veramente minuti e molto ben impaccati fra loro.
Nel campo della GASCROMATOGRAFIA le possibilità di scelta sono in un certo senso più limitate, soprattutto per via del fatto che la scelta fase mobile ha un peso decisamente minore (o non ne ha proprio) sui meccanismi di ritenzione, anche perché si tratta di un gas inerte che presenta solo una funzione di carrier, di trasportatore appunto, delle molecole in fase gassosa. Anche i parametri per la gestione fine del processo di separazione, ed in primo luogo la possibilità di lavorare in gradiente, non può giocare per lo stesso motivo sulla diversa composizione o concentrazione della fase mobile, e si concentrerà invece in primo luogo sulla variazione della temperatura, che costituisce uno dei fattori essenziali per gestire il processo di separazione gascromatografica.
La scelta della fase stazionaria in gascromatografia risente inoltre delle limitazioni imposte dalle elevate temperature alle quali sarà sottoposta in quello che non a caso viene definito come “forno” della colonna: a temperature superiori ai 300°C molti liquidi legati risultano essere tutt’altro che stazionari, e danno originre a notevoli rilasci, definiti bleeding della colonna.
Nei casi più semplici la fase stazionaria si comporta come un solvente per certi versi neutrale nei confronti delle sostanze da separare, limitandosi a solubilizzarle quando esse passano dallo stato aeriforme (trasportato nella fase mobile) allo stato condensato, tramite interazioni di Van der Waals, come nel caso del comune polidimetilsilossano (PDMS). L’ordine di eluizione in questo caso segue molto bene l’ordine dei punti di ebollizione delle sostanze da separare, tanto che la separazione cromatografica su queste fasi stazionarie simula in un certo senso il processo di distillazione frazionata.
In altri casi si introducono gruppi più complessi, per esempio parte dei metili sullo stesso silossano possono essere sostituiti con gruppi fenilici, con gruppi nitrilici, con ponti eterei e così via, e perché no anche gruppi con caratteristiche chirali, introducendo ulteriori meccanismi di interazione tra fase stazionaria e sostanze da separare che, come nel caso della cromatografia liquida, si possono liberamente sommare fra loro nel determinare l’ordine di eluizione ed il tempo di ritenzione dei diversi componenti della miscela iniziale.
Ma la limitazione più grande della separazione gascromatografica risiede probabilmente nel fatto che può essere applicata soltanto alle sostanze volatili, o comunque volatilizzabili alle temperature di esercizio. Per talune sostanze non volatili, come ad esempio i carboidrati o gli aminoacidi, è possibile “derivatizzare” le sostanze contenute nella miscela iniziale, ovvero sottoporre la stessa ad una reazione chimica con opportuni reagenti (derivatizzanti) in modo tale da trasformare le stesse molecole in altre più volatili e spesso anche meno reattive, solitamente tramite l’eterificazione o l’esterificazione dei gruppi più polari (es. ossidrile, carbossile, aminico, ecc) con gruppi trimetilsililicici, alchilici o acilici.
Nonostante questi limitazioni, la gascromatografia offre un paio di vantaggi di portata sufficiente a giustificarne l’affermazione in moltissimi campi applicativi: una capacità di risoluzione, ovvero di separazione fra sostanze chimiche anche estremamente simili fra loro, mediamente maggiore rispetto alle migliori tecniche di cromatografia liquida, e la possibilità di essere abbinate a sistemi di analisi in continuo della natura chimica delle molecole eluite, molto più descrittivi ed informativi di quelli disponibili per le tecniche di cromatografia liquida.
Il ruolo dei detector e l’impiego in chiave analitica piuttosto che preparativa o addirittura industriale delle tecniche di separazione cromatografica costituiranno l’argomento del successivo intervento sempre in tema di cromatografia.