Indice
Osservando gli atomi chimicamente legati fra loro all’interno delle molecole e forti del ricordo di come avevamo imparato fossero organizzati in un certo modo gli orbitali elettronici dei singoli atomi allo stato isolato non possiamo evitare di soffermarci un momento domandandoci se per caso non ci siamo persi qualche “passaggio intermedio” nel balzo concettualmente di certo non piccolo tra l’atomo e la molecola.
La configurazione elettronica di un atomo, la disposizione e la forma dei suoi orbitali di tipo s, p, d ed f intorno al nucleo e tutte le conoscenze che abbiamo messo insieme nelle giornate di studio precedenti, sembrano aiutarci ben poco a comprendere le ragioni per le quali le molecole siano fatte proprio così – e non in un altro modo – cioè con gli atomi legati in una certa posizione l’uno rispetto all’altro, con certo angoli e certe distanze di legame, fino a condizionare in modo determinante la forma della molecola stessa.
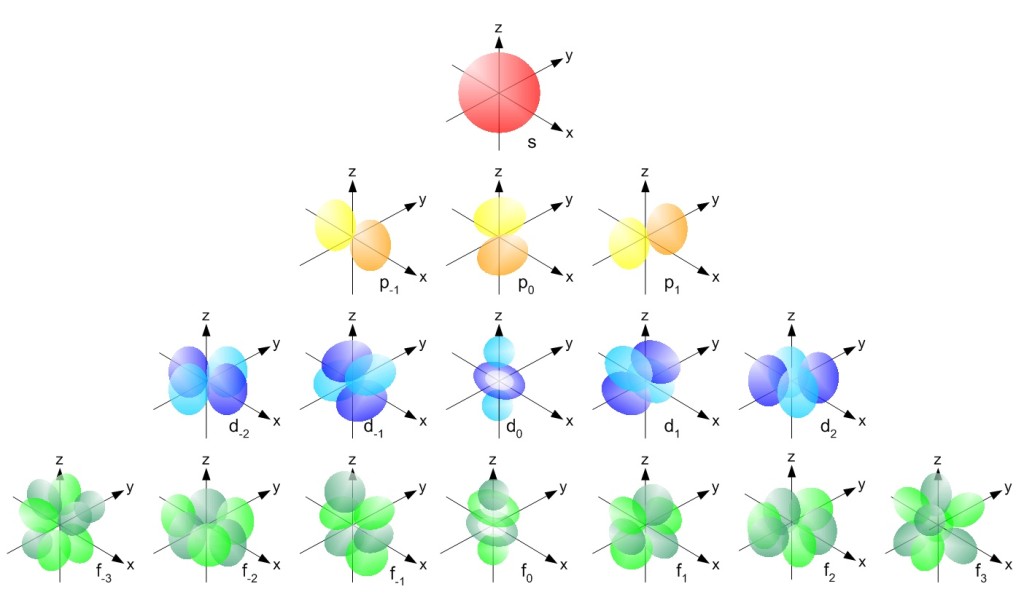
forma degli orbitali elettronici (s, p, d, f) dell’atomo isolato
Possiamo tentare di spiegarla in questo modo:
Nell’atto di stabilire legami chimici con altri atomi è particolarmente frequente che gli orbitali facenti parte del guscio elettronico più esterno di un atomo, molto simili fra loro dal punti di vista energetico ma diversi per geometria e per disposizione spaziale, si “combinino” fra loro con un processo noto come ibridizzazione – o ibridazione elettronica – restituendo un pari numero di orbitali atomici, questi detti ibridi, identici fra loro sia dal punto di vista energetico che della forma, molto ben direzionati nello spazio ed orientati a massimizzare la divergenza angolare reciproca all’interno dello spazio bi- o tridimensionale a loro disposizione. Il vantaggio energetico conseguente allo stabilirsi del legame chimico compenserà ampiamente la piccola promozione della quale possono godere talvolta alcuni elettroni per entrare a far parte degli orbitali ibridi, mentre la direzionalità specifica di tali orbitali costituisce un presupposto fondamentale per le caratteristiche geometriche e strutturali delle molecole che si formeranno, fino a rendere le stesse strutture molto meno “fluide” di quanto si sarebbe potuto immaginare in assenza del fenomeno dell’ibridizzazione.
1) (condizione) Gli orbitali atomici per combinarsi reciprocamente devono essere energeticamente simili fra loro. Questo significa in pratica che l’ibridizzazione può riguardare solo gli orbitali dello stesso livello energetico, ovvero con lo stesso numero quantico principale.
2) (condizione) L’atomo è impegnato a formare legami chimici con altri. In pratica l’ibridizzazione degli orbitali atomici riguarda i singoli atomi “in occasione” dell’instaurarsi di un legame chimico, e non atomi a sé stanti in forma non legata.
3) (conseguenza) Gli orbitali atomici di partenza sono sostituiti da un numero di orbitali (detti “ibridi”) numericamente uguale a quello degli orbitali che si sono combinati nell’ibridizzazione.
4) (conseguenza) Gli orbitali atomici formati – detti orbitali ibridi – sono fra loro identici per energia, intermedia rispetto a quella degli orbitali atomici che si sono fra loro combinati.
5) (conseguenza) Gli orbitali ibridi formati sono fra loro identici per forma, comunque più affusolata (ovvero maggiormente direzionati) rispetto agli orbitali di partenza.
6) (conseguenza) Gli orbitali ibridi formati sono disposti fra loro nello spazio tridimensionale secondo un criterio di ottimizzazione della loro divergenza angolare. Uno o più orbitali ibridi possono essere occupati da doppietti elettronici invece di partecipare attivamente ai legami molecolari.
7) (nota) L’ibridizzazione non è un fenomeno fisico ma una sorta di combinazione matematica… tra le funzioni d’onda che descrivono gli orbitali.
8) (nota) All’atto pratico, gli atomi che concorrono alla formazione di legami chimici – per lo meno a quelli covalenti – lo fanno tramite orbitali ibridi che coinvolgono tutti i in parte gli orbitali elettronici del livello elettronico più esterno.
9) (nota) L’ibridizzazione degli orbitali è parte integrante della teoria del Legame di Valenza e non viene considerata in contrapposizione con altre teorie pure in auge su argomenti analoghi (es. VSEPR e Teoria degli Orbitali Molecolari).
10) (applicazione) Si tratta di uno dei “metodi migliori attualmente disponibili” per rendere conto della direzionalità dei legami chimici e quindi della struttura delle molecole.
GLI ORBITALI IBRIDI DEL CARBONIO
Il fatto che l’ibridizzazione degli orbitali elettronici venga insegnata solitamente prendendo come esempio dell’atomo di carbonio non significa affatto che questo fenomeno non accada o abbia importanza minore per gli altri elementi.
Le ragioni per le quali il carbonio è certamente un buon esempio sono soprattutto tre:
– Si tratta di uno dei primi elementi della tavola periodica, uno dei più semplici;
– In esso si assiste anche ad un fenomeno collaterale, che spesso accompagna l’ibridizzazione degli orbitali: quello della promozione energetica di un elettrone dalle sue condizioni precedenti;
– La comprensione della chimica dei composti del carbonio, ovvero della chimica organica, ricava certamente grande beneficio dalla comprensione di una plausibile spiegazione della direzionalità dei legami, possibile previo ibridizzazione degli orbitali atomici dello stesso carbonio.
Come abbiamo detto l’ibridizzazione coinvolge orbitali energeticamente molto simili fra loro, quindi dello stello livello energetico – o con lo stesso numero quantico principale: dei 6 elettroni posseduti dall’atomo di carbonio isolato non consideriamo quindi i primi due, che occupano l’orbitale 1s ma ci concentreremo sui 4 rimanenti che occupano gli orbitali elettronici del livello 2.
Nel 2° livello i 4 elettroni non si trovano tuttavia uniformemente distribuiti fra i 4 orbitali possibili, ovvero il 2s ed i tre 2p diretti secondo le 3 coordinate spaziali (px, py, pz), bensì iniziano a riempire il sottolivello ad energia inferiore, quindi il 2s.
Nella figura a lato è rappresentata la configurazione elettronica del carbonio elementare allo stato atomico ed isolato.

Per potere ibridizzare i suoi orbitali elettronici 2s e 2p, uno degli elettroni presenti in coppia nel 2s dev’essere promosso a 2p: in questo modo si avrà una quaterna di orbitali (2s, 2px, 2py, 2pz) ciascuno occupato da un solo elettrone, che potranno fondersi reciprocamente restituendo 4 orbitali veramente identici per energia e per forma. Non essendoci ulteriori elettroni nella stessa fascia di distanza dal nucleo, questi orbitali potranno esprimere senza limitazioni la loro repulsione reciproca, ottimizzando la loro disposizione nello spazio tridimensionale. Se provate anche voi a collegare 4 bacchette identiche, libere di ruotare in ogni senso, ad una medesima pallina, vi accorgerete che c’è una ed una sola disposizione che consente di avere gli angoli fra due bacchette e la pallina al tempo stesso tutti uguali e maggiori possibile: quella a tetraedro.
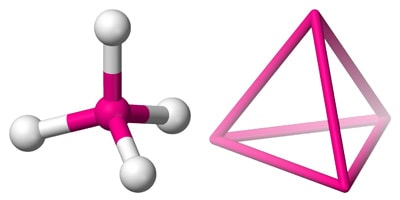
Il tetraedro è una figura geometrica di importanza assolutamente cruciale, che ricorre in natura ben più frequentemente di altre – giusto per fare un esempio il cubo – che pure per un motivo o per l’altro restano più facilmente incollate alla nostra memoria: basti dire che nessun bambino si ricorderà di aver giocato da piccolo con oggetti in plastica a forma di tetraedro e spesso prima dei 13-14 anni questa parola rischiamo di non averla neppure sentita nominare. Il tetraedro è in pratica una piramide a base triangolare dove ogni lato è costituito da un triangolo equilatero: in pratica quattro triangoli equilateri identici attaccati insieme per i lati. Quattro facce uguali, sei spigoli uguali e quattro vertici ed angoli uguali. In un’ibridazione sp3 l’angolo formato da due orbitali ibridi rispetto all’atomo centrale è di 109,28°.
L’atomo di carbonio che è andato incontro ad un’ibridizzazione sp3 viene così a trovarsi nel centro del volume di un tetraedro ideale, con i quattro orbitali ibridi diretti internamente verso i 4 vertici. Questo piccolo elemento strutturale tetraedrico dell’ibridazione sp3 del carbonio costituisce un tassello fondamentale dal quale parte l’edificazione di strutture organiche altamente complesse: lo ritroviamo nel metano, CH4, a legare quattro atomi di idrogeno, così come nel diamante a legare covalentemente altri quattro atomi di carbonio, che a loro volta ne legano altri quattro, e così via a loro volta, teoricamente all’infinito (salvo che il diamante ad un certo punto finisce). Anche il silicio, posto proprio un posto sotto, nello stesso gruppo del carbonio sulla tavola periodica ma nel periodo successivo, ovvero con il livello quantico 3 invece che con il 2 in corso di riempimento, gode normalmente di un’ibridazione di tipo sp3 in tutti i silicati, dove esso si pone al centro di tetraedri circondato da 4 atomi di ossigeno.
Il fatto che i 4 orbitali sp3, e di conseguenza i quattro legami che l’atomo di carbonio (e non soltanto) tetraedrico riesce a stabilire non possano “scivolare” l’uno sull’altro – cosa che comporterebbe, seppur temporaneamente, una sovrapposizione degli orbitali – ma restino al contrario confinati ciascuno nella propria zona con una certa rigidità, fa sì che quando questo atomo lega 4 atomi o gruppi differenti, siano possibili disposizioni fisse e fra loro diverse intorno ad esso, configurazioni non sovrapponibili tra loro ma che, al contrario, risultano essere l’immagine speculare l’una dell’altra. Anche in seguito a trasformazioni di tipo reattivo, sempre che la struttura tetraedrica non venga meno in nessun momento, gli atomi ed i sostituenti ai quattro vertici dell’atomo ibridato sp3 (in questo caso noto come centro stereogenico) possono cambiare, ma la configurazione intorno ad esso resterà la stessa.

Non necessariamente tutti gli orbitali elettronici originali di un atomo che “potrebbero” combinarsi per formare orbitali ibridi di fatto lo fanno.
Nel caso dell’atomo di carbonio, giusto per rimanere in tema, uno o anche due dei tre orbitali 2p possono restare fuori dall’ibridizzazione e comparire quindi in forma p originale nelle molecole formate da questo atomo di carbonio.
In particolare, dalla combinazione dell’orbitale 2s con 2 dei 3 2p si ottengono e orbitali sp2, obbligatoriamente schiacciati nello stesso piano a causa della perpendicolarità ad esso del restante orbitale atomico 2p che non si è combinato. E come per il tetraedro, qui il sistema migliore per uniformare e massimizzare gli angoli tra tre orbitali su di un piano è certamente quello di disporli a 120° l’uno rispetto all’altro.
L’orbitale p rimanente potrà contribuire alla formazione di un secondo legame, rendendo così “doppio” il legame semplice già stabilito dall’atomo di carbonio con uno dei suoi orbitali sp2: come conseguenza di questo, la morfologia dell’ibridizzazione sp2 e delle porzioni di molecole dove figurano atomi ibridati sp2 sarà di tipo piano.

Allo stesso modo, il triplo legame carbonio-carbonio corrisponde ad un’ibridazione sp (geometria lineare), che coinvolge un orbitale s ed un solo orbitale p per ciascuno dei due atomi di carbonio: i due orbitali atomici di tipo p, fra loro ortogonali, potranno quindi sovrapporsi lateralmente, andando a costituire il secondo ed il terzo legame, entrambe di tipo pi-greco.

orbitali sigma e pi-greco dell’acetilene
I DOPPIETTI ELETTRONICI
Non è raro trovare molecole neutre nelle quali due atomi legati ad uno centrale formino tra loro un angolo pressappoco di 120°, cosa che farebbe pensare ad un’ibridazione sp2 dell’atomo in questione… salvo il fatto che questo lega soltanto 2, e non 3 altri atomi come invece prevederebbe un’ibridazione di questo tipo.
Oppure un angolo di circa 109° tra due o tre direzioni di legame, come nell’ibridazione sp3… salvo il fatto che di atomi legati all’atomo centrale non ce ne sono 4, ma solo 3, o addirittura 2 soltanto.
Di fatto, uno o più di uno degli orbitali ibridi possono comportarsi come orbitali di non-legame, essendo occupati non da un elettrone passibile di condivisione, bensì di una coppia di elettroni, ovvero da quello che si è soliti definire un “doppietto elettronico”, in inglese ione pair. Seppur non contribuiscano ai legami chimici di tipo covalente più forte (come si vedrà di seguito i legami di tipo sigma), questi legami possono dare origine ad interazioni secondarie fra le molecole ma, cosa che in questo momento ci interessa sicuramente di più, condizionano in modo determinante la geometria della molecola, quasi come se si trattasse di orbitali di legame.
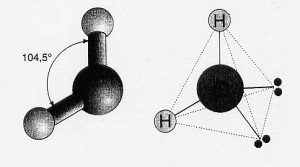
Un caso sotto gli occhi di tutti è quello della molecola dell’acqua nella quale, a differenza di quanto si potrebbe intuire, non vede i due atomi di idrogeno disposti lateralmente e sulla stessa linea rispetto all’atomo di ossigeno che li lega, ma angolarmente fra loro, formando un angolo H-O-H di 104,45°: la somiglianza con i 109,28° dell’angolo fra gli orbitali di un atomo ibridato sp3 non è casuale, perché è proprio da questa ibridizzazione che l’atomo di ossigeno parte nella molecola di acqua, subendo di seguito un ulteriore “schiacciamento” nell’angolo fra i due legami O-H a causa del fatto che i due orbitali ibridi che ospitano i due doppietti elettronici esercitano una repulsione elettrostatica maggiore rispetto a quella esercitata da due orbitali ibridi impegnati da legami chimici (nel legame con l’idrogeno, per esempio, almeno in parte gli elettroni condivisi gravano nell’ambito di competenza di quest’ultimo).
Un caso analogo è quello dell’ammoniaca, NH3, dove l’atomo di azoto risulterebbe “inspiegabilmente” posto al vertice di una piramide a base triangolare al posto che in centro ad un ipotetico triangolo equilatero circondato dai tre atomi di idrogeno come ci si potrebbe aspettare. Solo analizzando meglio la situazione si scopre che di fatto che l’azoto è all’incirca al centro di un tetraedro e che la posizione di vertice in apparenza libera è invece occupata da un doppietto elettronico, lo stesso che consente all’ammoniaca di esercitare il suo comportamento come base di Lewis.
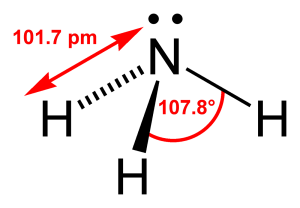
Molecole come queste, composte da atomi di elementi a diversa elettronegatività, disposti in modo asimmetrico (a causa della presenza di orbitali ospitanti doppietti elettronici) intorno ad un atomo centrale si comportano come dipoli elettrici. Una piccola peculiarità dalla quale dipendono un bel po’ di cose nell’universo, ad esempio il modo in cui la vita si è sviluppata e si mantiene ancora attualmente sulla Terra.
LEGAMI SIGMA E LEGAMI PI-GRECO
Nel linguaggio chimico si chiamano con la lettera greca σ (sigma) tutti i legami chimici dati dalla sovrapposizione “frontale” degli orbitali elettronici.
Nel caso degli atomi nei quali tutti o parte degli orbitali dello di valenza (quelli più esterni) risultano ibridizzati, i legami sigma sono dati dalla sovrapposizione frontale nel rapporto 1:1 di ciascun orbitale ibrido con un orbitale (ibrido o non) dell’atomo ad esso legato. I legami sigma si svolgono per tanto nella medesima direzione di estensione del legame ibrido e, seppur caratterizzati da notevole forza (dovuta ad un grande volume di sovrapposizione fra gli orbitali dei due atomi) essi lasciano i due atomi liberi di ruotare intorno al legame stesso. Questa possibilità di rotazione non viene solitamente percepita quando si trattano singoli atomi legati ma può portare a conseguenze anche molto importanti qualora l’atomo legato sia connesso ad una struttura molecolare più grande, con la conseguenza della possibilità di vari stati conformazionali corrispondenti ad angoli di rotazione differenti di una parte rispetto all’altra della molecola intorno ad un legame sigma.
Al contrario si chiamano π (pi-greco) i legami “ulteriori” che si possono eventualmente formare fra atomi già legati con legami sigma, per via della sovrapposizione degli eventuali orbitali p che sono rimasti esclusi dall’ibridazione. A differenza dei legami sigma, la sovrapposizione degli orbitali nei legami pi-greco è di tipo laterale. La duplice conseguenza di questo è da un lato quello di una forza inferiore rispetto al legame sigma (dovuta ad un volume inferiore di sovrapposizione fra gli orbitali dei due atomi), dall’altra una notevole rigidità del legame, che si oppone alla libera rotazione dei due atomi legati – e di conseguenza anche di eventuali porzioni di molecola – intorno al legame pi-greco.
Anche se il legame pi-greco è di per sé stesso più debole del sigma, dal momento che esso non si presenta da solo ma “si aggiunge” a due atomi già legati fra loro con legame sigma, il doppio legame complessivo risulta anche se non il doppio, comunque più forte rispetto al legame sigma da solo. Nel triplo legame, al quale prendono parte un sigma e due pi greco fra due coppie di orbitali p fra loro ortogonali, risulta essere un legame ulteriormente forte… ed anche un legame ulteriormente corto.

legami sigma e pigreco nella molecola di etilene
QUANDO ENTRANO IN GIOCO GLI ORBITALI D: LA GEOMETRIA DEI COMPLESSI DI COORDINAZIONE
Non solo organica, non solo carbonio e non solo covalenti i legami che possono prendere forma – o meglio, per giocare un po’ con le parole, “orientamento” – dall’ibridazione degli orbitali elettronici.
Gli elementi di transizione infatti alle già descritte possibilità di ibridazione fra orbitali s e p, possono entrare in gioco con i loro caratteristici orbitali d, rendendo così possibili un gran numero di differenti opzioni come ad esempio: sd, sd2, sd3, sd4, sd5, sp3d, sp3d2.
Anche semplicemente dall’espressione del tipo di ibridizzazione riusciamo già a leggere fra le righe il numero di orbitali fra loro isomorfi ed isoenergetici che si formeranno (ad es. 5 per l’sd4, 6 per l’sp3d2, ecc) e quindi, per le note ragioni di organizzazione spaziale, anche la disposizione dei legami – o meglio dei loro angoli reciproci – intorno all’atomo ibridato, e quindi anche la geometria dei sostituenti.
Proseguendo l’esame delle figure geometriche già affrontato per le ibridazione sp, sp2 ed sp3, quelle che comportano 5 legami sigma, come ad esempio la sd4 e la sp3d definiscono con la direzione dei propri orbitali ibridi i vertici di una bipiramide trigonale e quelle che comportano 6 legami sigma, come ad esempio sd5 e sp3d2 formano invece un ottaedro, ovvero una bipiramide tetragonale.
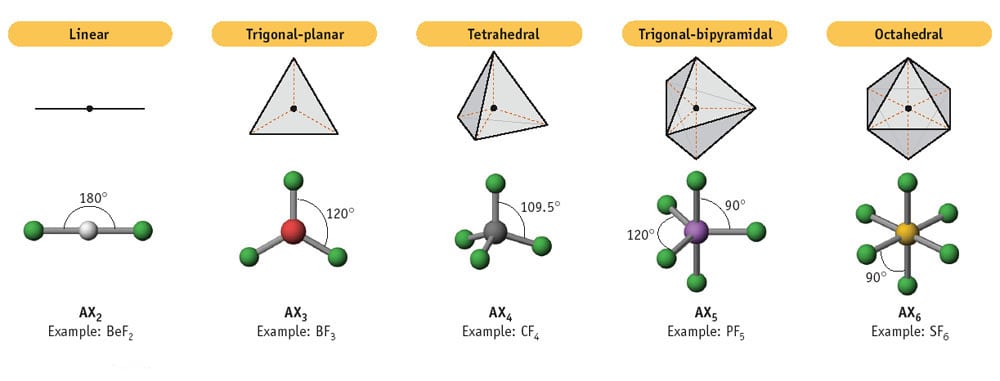
Queste geometrie si riscontrano con grande frequenza nei complessi di coordinazione che caratterizzano appunto gli elementi di transizione del blocco d della tavola periodica, dove un atomo del metallo di transizione i cui orbitali elettronici esterni risultano variamente ibridati fra loro, lega con legami sigma molecole neutre in grado di fornire doppietti elettronici come base di Lewis (es. acqua, ammoniaca, ossido di carbonio, ecc) o anioni (idrossido, alogenuri, nitrito, ecc) formando così strutture complesse a carattere neutro o ionico, spesso anche solubili in acqua, ancora più spesso intensamente colorate.
Ne sono degli esempi il [Cu(NH3)4(H2O)2]2+ dal colore blu quasi violaceo molto intenso, stabile ovviamente solo in soluzione e solo in ambiente basico, che si può ottenere per graduale basificazione di una soluzione acquosa di un sale di rame(II) con soluzione acquosa di ammoniaca, ma in generale un po’ tutte le forme assunte dai sali più o meno idratati dei metalli di transizione, presenti in soluzione acquosa neutra per lo più come acquacomplessi.
La variazione da uno stato di idratazione ad un altro dell’atomo di cobalto, con geometria differente nella disposizione delle molecole di acqua ad esso legato, sta alla base della variazione di colore del cloruro di cobalto cristallizzato, dalla forma anidra (blu) a quella diidrata (viola) a quella esaidrata (rosa): questa variazione di colore è utilizzata tanto negli indicatori colorimetrici di umidità (ad es. in abbinamento con il gel di silice negli essiccatori) che su quelle statuine in vero un po’ kitsch che cambiano colore con il variare delle condizioni di umidità dell’aria.

Analogamente al ruolo stereogenico del carbonio ibridato sp3 nelle molecole chirali, anche l’elemento di transizione impegnato in complessi di coordinazione con l’impiego degli orbitali d ibridati, a parità qualità e quantità di atomi legati può dare origine a configurazioni diverse, distinte sulla base delle posizioni reciproche di questi atomi l’uno rispetto all’altro e rispetto all’atomo centrale.
Il complesso di formula [CoCl2(NH3)4]+, per esempio, caratterizzato da 2+4=6 orbitali ibridi identici fra loro e quindi presumibilmente da una forma ottaedrica, può portare i due sostituenti Cl vicini fra loro, ovvero con un angolo Cl-Co-Cl di 90° (configurazione detta “cis”), oppure in direzione opposta, sulla stessa linea rispetto all’atomo centrale, ovvero ai due vertici dell’ottaedro (configurazione “trans”). La stessa differenziazione vale nel caso del complesso neutro [CoCl3(NH3)3] dove i tre atomi uguali possono essere disposti sullo stesso piano in comune con l’atomo centrale di cobalto oppure no.
Sempre a condizione che i complessi siano sufficientemente forti nei loro legami di coordinazione, ovvero che i sostituenti non siano liberi di staccarsi e ricongiungersi all’atomo centrale come accede per i legami a carattere strettamente ionico, le diverse conformazioni assegnabili a parità di formula bruta ad un composto di coordinazione corrispondono di fatto a specie chimiche differenti, singolarmente sintetizzabili ed isolabili, dotate di caratteristiche chimico-fisiche talora diverse. D’altronde si tratta anche in questo caso di coppie, o addirittura di serie, di stereoisomeri.

TEORIE E PUNTI DI VISTA
Questo titoletto suonerà certamente come una sgradevole provocazione per chi coltiva il mito della scienza come arbitro ultimo ed univoco della coerenza della natura.
In realtà la Natura è chiara e coerente nel suo comportamento: è la sua interpretazione, ovvero la ricerca delle relazioni causali nel suo comportamento – un’esigenza ben inteso tutta umana – ad esserlo assi meno. E’ così che nel corso della storia della scienza vengono formulate ipotesi, spiegazioni e talvolta vere e proprie teorie per spiegare alcuni aspetti della Natura che, seppur non si dimostrino “false” a distanza di anni, possono tuttavia essere sostituite da altre teorie ancora più soddisfacenti, o evolvere o sfociare in quadri di conoscenza più ampi.
Nell’ambito della struttura della materia – e quello che stiamo trattando ora ne è un caso – per descrivere in fondo lo stesso argomento sono state formulate teorie diverse, a distanza di anni o talvolta quasi in contemporanea, ognuna delle quali ha dei pregi e dei difetti. Nel migliore dei casi una riesce a spiegare bene alcuni aspetti dell’argomento, ad esempio alcuni comportamenti della materia, dove l’altra teoria sembra annaspare di più; in altri casi semplicemente una teoria vanta basi matematiche più rigorose e dimostrabili passo per passo ma risulta di più ostica applicazione (ad esempio richiederebbe apparati di calcolo al momento imensabili) mentre l’altra, seppur meno rigorosa si presta ad una applicazione più intuitiva riuscendo a spiegare meglio i comportamenti osservati e/o a prevederne di nuovi.
E in fondo, non è a questo che ci serve la conoscenza, specie quando prende la forma di una teoria?
Il ricorso concettuale ai legami di tipo sigma e di tipo pi greco per giustificare caratteristiche geometriche ed energetiche dei legami chimici e, di conseguenza, delle molecole nelle quali essi figurano, non dipende conseguenzialmente dalla teoria dell’ibridazione degli orbitali atomici, essendo fra l’altro storicamente precedente ad essa. E’ dagli anni ’20 del XX secolo, infatti, che la Teoria del Legame di Valenza (VBT) formulata da Linus Pauling, W. Hitler, F. London e J. Slater ha introdotto i concetti di legame sigma e pi greco insieme al concetto di fondo secondo il quale i legami chimici si formerebbero come conseguenza dell’appaiamento degli elettroni degli orbitali dello strato di valenza degli atomi, attraverso quella che viene solitamente interpretata come una fusione per sovrapposizione di questi orbitali atomici.
L’ibridizzazione degli orbitali elettronici di tipo atomico è stata di fatto introdotta successivamente, a partire dallo stesso Pauling come evoluzione della suddetta Teoria del Legame di Valenza, nel suo noto libro pubblicato nel 1939 ed ancora oggi largamente ristampato con l’introduzione ed i commenti da parte di illustri chimici The Nature of the Chemical Bond.
Il ricorso all’ibridizzazione degli orbitali atomici introdotta, seppur ancora in sordina in occasione di questa pubblicazione, si è subito affermata grazie alla sua “usabilità” ai fini della spiegazione e della previsione della geometria di ioni e molecole, nonché talvolta addirittura dello stato di valenza effettivamente dimostrato da alcuni elementi, ad iniziare dallo stesso carbonio degli esempi riportati nei capitoli precedenti (es. il metano sarebbe stato una struttura inspiegabile tenendo conto della sola teoria del legame di valenza).
E’ attualmente in corso un dibattito scientifico relativo non tanto alla fondatezza scientifica, quanto all’utilità ed al valore specie in sede didattica delle diverse teorie attualmente accettabili per spiegare ed interpretare le caratteristiche geometriche ed energetiche dei legami chimici e quindi delle molecole delle quali essi fanno parte.
A questo proposito si sta affermando sempre più la proposta, almeno a livello di percorso di studi non specialistico, di evitare il ricorso certamente più complesso dal punto di vista delle comprensione teorica ed in questa sede tutto sommato poco “utile” della Teoria degli Orbitali Molecolari (MO) ma anche di quella del Legame di Valenza con la sua evoluzione in funzione dell’ibridizzazione degli orbitali atomici, a favore invece della Teoria VSEPR (Valence Shell Electron Pair Repulsion, ovvero repulsione delle coppie elettroniche nel guscio di valenza) che, seppur di più immediata comprensione, avrebbe il difetto di mal adattarsi ad un’applicazione previsionale di tipo quantitativo.