Un profumo, un olio essenziale, un frutto aromatico: quello che il nostro naso percepisce odorando un fiore o spesso anche semplicemente assaggiando un alimento, come molti sicuramente sanno, è solitamente un insieme di decine, per lo più centinaia di molecole diverse, in differenti rapporti quantitativi fra loro.
L’approccio conoscitivo tipico per descrivere in modo univoco la complessità di un profumo, nonché per riuscire successivamente a riprodurlo, è quello di tradurre la sua composizione in una stringa numerica o meglio ancora una matrice bi- o tridimensionale.
Per fare questo dobbiamo utilizzare in primo luogo una tecnica di risoluzione che separi i singoli componenti (molecole) della miscela complessa (1° dimensione della matrice) e su ogni componente separato intervenire con una tecnica analitica che fornisca un responso coerente con la struttura della molecola separata (2° dimensione).
Mi spiego meglio.
Sui metodi di separazione a cui sto pensando, che vanno sotto il nome generico di “cromatografia”, si tengono interi corsi universitari, girano intere aziende di strumentazione scientifica ed esistono decine di riviste specializzate. Non me ne vogliano quindi gli esperti nel settore (io stesso utilizzo queste tecniche da circa 15 anni!) se in questa sede semplifico il tutto dicendo che la cromatografia è in ultima istanza un metodo per separare una miscela nei suoi componenti sulla base del tempo.
Concettualmente e del tutto al di là di come questo possa accadere, io fornisco in input allo strumento una miscela di molecole (quello che per noi è ad esempio un’essenza) tutte insieme, ed in output lo strumento le molecole usciranno singolarmente in un certo arco temporale. Ci saranno istanti nei quali non uscirà alcunché, istanti nei quali uscirà una specifica molecola1, poi una specifica molecola2 di tipo differente (ovvero un’altra sostanza chimica) e così via, fino alla separazione nel tempo di tutti i diversi componenti chimici della miscela.
Se all’uscita di questo separatore pongo un analizzatore genericamente sensibile alla perturbazione del suo stato di quiete, questo fornirà un segnale, facilmente traducibile in un segnale elettrico, ogni qual volta una molecola uscirà dal separatore cromatografico. Un analizzatore generico di questo tipo può essere definito di tipo “bulk” in quanto non fornisce informazioni sulla natura della molecola che gli sta passando davanti ma fornisce semplicemente un segnale proporzionale alla sua concentrazione.
L’oggetto matematico generato da un accoppiamento costituito da un cromatografo + un detector bulk è una stringa, dove la dimensione lineare è rappresentata dalla scansione temporale (di solito una ogni frazione di secondo, a fronte di un tempo analitico complessivo di almeno qualche minuto) mentre il valore assunto da ogni cella è un numero positivo proporzionale alla concentrazione della specie chimica. Il tutto può essere rappresentato su un diagramma cartesiano bidimensionale, con in ascisse il tempo ed in ordinate l’intensità del segnale elettrico registrato: una rappresentazione del genere prende il nome di cromatogramma ed ogni picco è relativo ad una diversa sostanza chimica contenuta nel nostro profumo come in qualsiasi altra miscela di sostanze chimiche. La forma ideale del picco è una gaussiana, ma esistono un bel po’ di fenomeni di natura fisica e chimica che concorrono nel minare questa aspettativa teorica, per lo più rendendo asimmetriche le code con una particolare allungamento frequente specie nella parte discendente della curva, ovvero verso i tempi maggiori.
Sempre rimanendo in ambito di calcolo, la concentrazione di ogni componente è proporzionale all’integrale definito sotteso fra la curva del segnale e la linea di base, ovvero quella che si può ottenere congiungendo i punti del grafico corrispondenti agli istanti nei quali non usciva alcun componente dal sistema cromatografico, in pratica il rumore di fondo. La concentrazione di ogni componente diventa in questo caso proporzionale alla differenza degli integrali fra le due curve ed integrato sulla scala dei tempi fra l’inizio e la fine del picco.
Il fattore di proporzionalità di solito è specifico per ciascuno tipo di molecola, dipende inoltre dalla tecnica di detezione prescelta e solitamente lo si può calcolare partendo da uno standard, ovvero dalla stessa sostanza chimica, utilizzata a due o più concentrazioni diverse: tabulando le concentrazioni d’impiego contro l’area netta integrata del picco riferito a quella specifica sostanza, si otterrà una retta e la pendenza della retta costituisce il fattore di proporzionalità fra concentrazione ed area integrata.
Questo fattore è valido in tutto il range definito “di linearità”, specifico per ogni assetto analitico adottato (cromatografia+detector): se dovessimo utilizzare concentrazioni troppo ridotte della sostanza rischieremo infatti di sconfinare al di sotto del LOQ (limite inferiore di quantificazione) dove il picco analitico si confonde matematicamente con il rumore di fondo, mentre a concentrazioni troppo alte rischieremo di saturare il segnale, con il raggiungimento di un fattore di riposta via via decrescente fino ad un possibile plateau.
Una tecnica analitica ottimale sfrutta un range di linearità di almeno un paio di ordini di grandezza di concentrazioni di analita (nome attribuito alle molecole che vengono ricercate nell’analisi), e se questo risulta eccessivamente concentrato si provvede a pre-diluirlo prima di sottoporlo all’analisi.
Capita talvolta, specie nelle miscele più complesse, che la tecnica cromatografica utilizzata non sia in grado di separare perfettamente nel tempo due componenti chimici molto simili fra loro. Il picco che si osserva in questi casi risulta sdoppiato, con due punte oppure con una piccola spalla a sinistra o a destra. In questi casi si utilizzano algoritmi particolari di deconvoluzione per ricostruire l’equazione algebrica della curva che dovrebbe teoricamente avere ciascuno dei componenti nella zona di sovrapposizione, solitamente cercando di ricorrere all’approssimazione ottimale di una curva gaussiana, e l’integrazione viene pertanto calcolata su queste nuove curve.
Ma a questo punto ancora nulla sappiamo della reale identità dei singoli componenti che abbiamo separato e tracciato sul cromatogramma. L’approccio in molti casi più semplice può essere quello di procurarsi degli standard di riferimento, ovvero un set di alcune delle molecole che potrebbero risultare fra le migliori candidate alla presenza in quello specifico campione, e sottoporle singolarmente allo stesso identico procedimento di separazione e detezione: se il tempo esatto di uscita (detto tempo di ritenzione cromatografia) è il medesimo di quello di uno dei picchi sul cromatogramma complesso del campione, ecco che potremmo affermare che si tratta della stessa sostanza chimica. In un sistema ideale infatti e a parità di condizioni di trattamento, il tempo di ritenzione è una variabile numerica derivata da fattori molteplici ma tutti univocamente dipendenti dalla struttura della molecola, ovvero dalla tipologia di atomi, dal loro numero e soprattutto dalle loro modalità di legame reciproco.
L’alternativa migliore resta però quella di utilizzare per l’analisi non un detector generico ma un detector “descrittivo”, in grado di generare un’informazione complessa relativamente ad ogni componente in uscita dal separatore cromatografico. Per esempio ad intervalli discreti di tempo, di solito comunque sull’ordine della frazione di secondo, questo tipo di detector esegue una scansione di una proprietà fisica ciclicamente variabile ed allo stesso tempo, ad ogni ciclo, registra il comportamento da parte di ciò che in quell’istante sta uscendo dalla separazione. Fra le proprietà fisiche più comunemente misurate risultano soprattutto la capacità di interagire con radiazioni elettromagnetiche, solitamente nel dominio dell’ultravioletto, del visibile o dell’infrarosso: se ad ogni frazione di secondo un sistema automatico sarà in grado di eseguire in uscita al separatore cromatografico una veloce scansione spettrale da una data lunghezza d’onda ad una data altra, un rivelatore posto a seguito potrebbe essere in grado di misurare, in modo altrettanto rapido, gli smorzamenti percentuali di intensità (rispetto all’originale emesso dalla sorgente) di questa radiazione in funzione della sua lunghezza d’onda. Quello che si otterrà sarà uno “spettro di assorbimento” ed anch’esso come il cromatogramma può essere rappresentato come una stringa, dove la dimensione lineare è costituita dalle lunghezze d’onda verificate (solitamente, per quanto vicine, si tratta di valori discreti), mentre il valore assunto da ogni cella è proporzionale a quanto lo specifico componente chimico separato è in grado di assorbire a quella determinata lunghezza d’onda. Riportato su un diagramma, lo spettro di assorbimento riporterà sull’asse delle ascisse un valore proporzionale all’energia della radiazione (es. lunghezza d’onda oppure frequenza oppure numero d’onda), mentre sull’asse delle ordinate comparirà un valore proporzionale all’assorbanza o al concetto ad esso complementare, ovvero la trasmittanza.
Lo spettro di assorbimento elettromagnetico, sempre a parità di condizioni di ottenimento, è una preziosa informazione descrittiva della molecola a cui si riferisce.
Immaginando di ripetere la scansione spettrale ad intervalli vicinissimi di tempo, si arriverà a costruire una seconda dimensione analitica: quelle che una volta erano semplici caselle di una stringa cromatografia, dipendenti dal tempo, diventeranno ora intere righe e una matrice così prodotta avrà dimensioni tempo x lunghezza d’onda, con i valori numerici riportati nelle celle corrispondenti all’assorbimento ad un certo tempo per una certa lunghezza d’onda.
Una delle tecniche di analisi spettrali più potenti in termini di potere discriminante è la spettrometria di massa. In questa tecnica la molecola che in quell’istante esce dal separatore cromatografico viene ionizzata e spezzata in più frammenti anch’essi dotati di carica elettrica (comunque tutte coerenti nel segno, diverso a seconda delle tecniche adottate), ed anche i frammenti potranno essere spezzati in sottoframmenti: la cosa interessante è che a parità di condizioni operative, il percorso di frammentazione e quindi gli aspetti quali- e quantitativi della collezione di frammenti carichi che si otterrà è una proprietà altamente caratteristica della molecola di origine, tanto che (almeno nel caso della sottotecnica utile nella definizione dei profumi) sono disponibili commercialmente database elettronici di spettri di massa costituiti da più di 600000 spettri di diverse molecole, con algoritmi di comparazione molto raffinati che consentono in meno di un secondo di individuare fra tutti lo spettro che meglio fitta con quello sperimentale.
Abbiamo quindi ottenuto una matrice visualizzabile anche per mezzo di plot tridimensionali, anche se il chimico preferisce “affettare” il tutto secondo una delle due dimensioni: estraendo una riga a tempo costante otterrà lo spettro di massa per un singolo componente (o per il rumore di fondo nel caso in quell’istante di fronte al detector di massa non transitasse nessun componente in uscita dal separatore cromatografico); estraendo una colonna a massa costante (o lunghezza d’onda nel caso di detector a scansione di radiazione elettromagnetica) si otterrà invece la distribuzione di quella specifica massa in relazione al tempo, evidenziando ad esempio in quali componenti chimici essa si distribuisce ed in che rapporto.
Creando un’ulteriore riga dove saranno calcolate le sommatorie dei valori assunti dalle varie celle, colonna per colonna, si otterrà la riga del TIC (Total Ion Chromatogram) che ha lo stesso significato della stringa ottenuta tramite un analizzatore di tipo bulk.
Se le scansioni spettrali saranno sufficientemente rapide da poter essere eseguite più volte nell’arco dell’uscita di un singolo componente dal separatore cromatografico, la sua gaussiana teorica, ovvero il suo picco, potrà essere suddiviso in intervalli temporali discreti e su ciascuno di essi sarà possibile osservare un diverso spettro di massa. Se le diverse scansioni sullo stesso picco dovessero differire in modo significativo nei rapporti relativi fra le diverse masse in essi contenute, se ne dedurrà che il componente in uscita non era puro, bensì quello che vedavamo come un picco cromatografico unico era in realtà dato dalla sovrapposizione di più componenti: la sottrazione algebrica degli spettri di massa fra scansioni diverse eseguite sullo stesso picco sarà in grado di fornire gli spettri dei diversi componenti puri, singolarmente ricercabili nei suddetti database spettrali per un’attribuzione qualitativa della tipologia di molecola in questione.
Alla fine, ottenuta la collezione completa di spettri di massa puri registrabili a partire da un campione iniziale sottoposto a separazione cromatografica, si può dire di avere a disposizione l’elenco, quali- e volendo anche quantitativo, dei componenti chimici contenuti in quel dato campione.
Le matrici ottenute in questo modo costituiscono un ottimo modello per descrivere nei dettagli la composizione di un prodotto complesso analizzabile, ed in modo specifico di un profumo. Si noti che alcune proprietà del calcolo matriciale non sono in questo caso direttamente utilizzabili: il concetto di sommatoria per esempio dovrebbe essere costituito da quello di media in quanto subentra quello che di può definire “fattore di diluizione”. Semplificando il discorso a livello di stringa, senza nulla perdere nell’estensione a matrice, ad esempio, si noterà che miscelando quantità uguali di un campione 2; 0; 4; 3; con un campione 0; 2; 6; 3 si otterrà non un campione 2; 2; 10; 6; bensì la media fra i due: 1; 2; 5; 3.
Recentemente si stanno iniziando ad ultizzare sistemi analitici dove ulteriori dimensioni sono introdotte, sia sul versante cromatogafico che su quello della detezione. Sul versante cromatografico per esempio è stata introdotta la cromatografia bidimensionale: ogni qualche secondo il prodotto in uscita dal primo separatore cromatografico viene introdotto automaticamente in un secondo, più veloce, in grado di terminare la separazione su base tempo a sua volta nell’arco di pochi secondi.
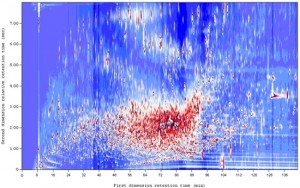
La matrice che si ottiene conterrà così righe relative alla separazione esercitata su base tempo dal primo sistema: ciascuna cella viene poi “esplosa” nell’altra dimensione in relazione alla separazione esercitata dal secondo separatore cromatografico; la terza dimensione sarà così relativa alla scansione degli spettri di massa e la quarta… si comprende facilmente come un sistema con questo grado di complessità risulti difficilmente rappresentabile e sicuramente ben poco utile, se non appunto previa filtrazione su un plot semplicemente bidimensionale, dove il colore viene solitamente utilizzato per rappresentare la terza dimensione.
La conoscenza di queste matrici composizionali e delle possibilità di trattamento numerico ad esse applicabili possono tornare utili per esempio nella ricostruzione di uno specifico profumo del quale non si conosca la composizione:
dovremmo innanzitutto disporre di un database ben fornito di matrici composizionali delle principali materie prima potenzialmente utilizzabili, quindi applicare il processo analitico esaminato anche sul profumo oggetto di interesse. Si può immaginare di ricostruire il profumo d’interesse (temine noto) come combinazione lineare di tutte le materie prima, singolarmente, dove i coefficienti dell’equazione costituiscono i “pesi”, ovvero i rapporti relativi di miscelazione (quindi zero se il componente non è da aggiungere).
Per ottimizzare questa miscela dovrebbero essere trovati numerosi coefficienti per un’unica equazione, senza la possibilità purtroppo di poterla mettere a sistema con altre equazioni.
Il sistema più comunemente utilizzato è quello di utilizzare un algoritmo su PC in grado di generare numeri da principio casuali da sostituire ai vari coefficienti, ed affinare via via la ricerca qualora i due termini dell’equazione, quello noto che rappresenta il profumo da simulare e quello di calcolo che rappresenta la simulazione, dovessero iniziare ad avvicinarsi fra loro: ovviamente l’eguaglianza perfetta fra i due membri è tutt’altro che garantita, ma almeno si potrà dire di essersi avvicinati il più possibile alla realizzazione di un piccolo miracolo che fino a pochi anni fa era privilegio di pochi, espertissimi profumieri.