Indice
Risulta essere molto importante prendere subito coscienza che i metodi che applicheremo per il calcolo del pH nelle diverse situazioni, sono tutti metodi approssimati. Ovvero si basano su determinate assunzioni che si fanno di volta in volta per semplificare la natura del calcolo, che altrimenti potrebbe risultare assai complesso e laborioso. Le approssimazioni introdotte sono lecite a condizione che il risultato non sia significativamente diverso rispetto al calcolo “esatto”. In pratica, sono accettabili soluzioni che contengano un “errore” non superiore all’1% (sul pH) rispetto al calcolo “esatto”. L’approssimazione che si fa più di frequente riguarda il contributo dell’autoionizzazione dell’acqua alla concentrazione di H+ (o di OH- ), di cui si deve tener conto solo nel caso di soluzioni molto diluite. L’espressione “molto diluite” deve comunque essere rapportata alla forza dell’acido (o della base): più l’acido (o la base) è debole, più “critico” diventa il fattore diluizione. Un altro genere di approssimazione si fa spesso nel caso di un’unica specie monoprotica, ogni volta che la concentrazione dell’acido (o della base) all’equilibrio si considera uguale alla sua concentrazione analitica. Questa approssimazione è lecita solo per acidi (o basi) effettivamente deboli, e, come vedremo, a condizione che la soluzione non sia eccessivamente diluita. In questo caso, l’approssimazione è quindi legata sia al valore di Ka (o Kb), sia alla concentrazione dell’acido (o della base). Infine, nel caso di specie poliprotiche o anfotere, le ulteriori approssimazioni sono essenzialmente basate sui valori relativi delle varie Ka e/o Kb. Questi concetti e la natura delle approssimazioni verranno chiariti caso per caso.
Prima di affrontare il problema del calcolo del pH, dovreste aver ben chiaro il concetto di acido e base, visto attraverso la teoria di Bronsted e Lowry. È infatti preferibile acquisire dimestichezza con le reazioni in cui questi composti sono coinvolti e, soprattutto, comprendere i concetti che sono alla base dell’equilibrio chimico, piuttosto che imparare a calcolare il pH “esatto” di una soluzione di acido solforico.
Il pH di Acidi e Basi forti
Si definiscono acidi forti quegli acidi che in soluzione sono completamente dissociati. Sono acidi forti tutti gli idracidi degli alogeni (ad eccezione del Fluoro), e gli ossoacidi a più alto grado di ossidazione dell’Azoto, dello Zolfo, degli alogeni (tranne ancora il Fluoro) e del Manganese. In accordo con la teoria di Arrhenius, la loro dissociazione può essere indicata come, ad esempio:
HCl => H+ + Cl–
In accordo con la teoria di Bronsted e Lowry si scriverà invece:
HCl + H2O => H3O+ + Cl–
Formalmente, H+ e H3O+ indicano la stessa sostanza e sono quantitativamente equivalenti.
Fu Lowry il primo ad introdurre il concetto di ione ossonio.
L’unica freccia che compare in queste reazioni ha lo scopo di sottolineare che gli equilibri sono completamente spostati a destra. Ciò significa che la concentrazione di H+ e H3O+ è praticamente uguale alla concentrazione cosiddetta analitica dell’acido, ovvero a quella quantità di acido che è stata posta in soluzione.
![]()
Un’ulteriore concetto che scaturisce dalla teoria di Bronsted e Lowry è quello del cosiddetto effetto livellante del solvente. In sintesi, il solvente riduce l’acido più forte in soluzione alla forma protonata del solvente stesso; se il solvente è l’acqua, l’acido più forte che possa esistere in soluzione è lo ione H3O+ . Per le sue proprietà anfiprotiche, l’acqua ha un effetto livellante anche sulle basi forti: la base più forte che possa esistere in soluzione acquosa è lo ione OH–.
Basi Forti
Per affrontare il calcolo del pH di una soluzione di una base forte, sarà sufficiente utilizzare la teoria di Arrhenius per definire basi forti quelle sostanze che in soluzione si dissociano completamente in ioni del metallo e ioni OHGli idrossidi sono dunque tipiche basi forti. Tuttavia occorre fare attenzione a non “assolutizzare” questa affermazione. Con gli idrossidi dei metalli alcalini non ci sono problemi. Se ad esempio vi viene chiesto di calcolare il pH di una soluzione 0.1 M di idrossido di sodio, sulla base della definizione che abbiamo appena dato, scrivete la reazione
NaOH => Na+ + OH–
la quale indica che la [OH-] è uguale alla concentrazione analitica della base
forte, C°b. Quindi
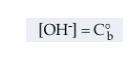
Si calcola quindi il pOH (=1) e infine il pH da pH = 14 – pOH = 13.
Nel caso degli idrossidi dei metalli del II gruppo (e non solo), occorre fare attenzione al fatto che alcuni non sono completamente solubili, ma sono caratterizzati da un certo valore del loro “prodotto di solubilità”. Attenzione quindi a non cadere in certi automatismi, quali quello di considerare la [OH-] doppia rispetto alla concentrazione dell’idrossido.
Idrossidi e Prodotto di solubilità
Frequentemente, gli idrossidi vi sono stati presentati come basi forti per eccellenza. Tuttavia, non pochi di essi, tra cui alcuni dei metalli del II gruppo (Mg e Ca) e molti dei metalli di transizione (Cr, Mn, Fe, Co, Ni, Zn, Cd) sono poco solubili. Tanto che la definizione va attentamente riconsiderata.
Quando si scioglie un sale nell’acqua (anche gli idrossidi possono essere considerati tali) prima o poi ci accorgiamo che, continuando ad aggiungere il sale, questo fa fatica a sciogliersi completamente ed inizia a formarsi un “corpo di fondo”. A quel punto, la soluzione si dice satura e gli ioni del sale hanno raggiunto in soluzione la loro massima concentrazione possibile. Il valore della concentrazione del sale in soluzione è detto solubilità e può variare, anche parecchio da sale a sale. Alcuni sali, quali nitrati, clorati, molti alogenuri (specie dei metalli del I gruppo) sono generalmente così solubili che si possono considerare “completamente solubili”. Altri sono invece scarsamente solubili (molti solfati, solfuri, carbonati, idrossidi, etc.) e la loro solubilità è definita attraverso il cosiddetto prodotto di solubilità, Kps.
Il Kps è praticamente la costante di equilibrio che si applica, in conformità con la legge di azione di massa, alla reazione di dissociazione del sale, in una sua soluzione satura (si tenga presente che questa precisazione è essenziale). Per esempio, per
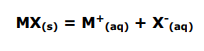
si ha
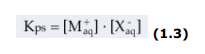
La concentrazione del sale “solido” si considera convenzionalmente = 1. Eventuali coefficienti stechiometrici sono posti a esponente nell’espressione di Kps, esattamente come si fa con una costante di equilibrio, nell’applicazione della legge di azione di massa.
Per esempio, per un idrossido di un metallo del II gruppo, la cui dissociazione è descritta dalla reazione
![]()
Kps è
![]()
Qual è il significato dell’espressione 1.3?
La Kps può essere interpretata come una condizione “limitativa”, nel senso che ci permette di affermare che in una qualsiasi soluzione acquosa del sale, il prodotto di [M+ ] x [X– ] deve essere inferiore, o al massimo può essere uguale, a Kps. Nel caso di un idrossido poco solubile, Me(OH)n, se la sua concentrazione analitica è tale che il prodotto [Men+] x [OH– ] n è minore di Kps, l’idrossido in soluzione è completamente dissociato; viceversa, se il prodotto è maggiore di Kps, significa che si è superata la “soglia di solubilità”, e nella soluzione è presente il corpo di fondo, ovvero una certa quantità di idrossido “solido”.
Come si calcola la soglia di solubilità?
Facciamo un esempio concreto, scegliendo l’idrossido di calcio (Kps =5.5 x 10-6) come cavia. Si ponga [Ca2+] = x; la [OH–] sarà di conseguenza 2x. Dobbiamo quindi risolvere l’equazione
![]()
da cui, x = 0.011 moli/litro, che rappresenta la [Ca2+] e anche la solubilità del sale.
Fino a questa concentrazione, l’idrossido in soluzione è completamente dissociato (la [Ca2+] = [idrossido] e la [OH–] = 2*[idrossido]), per concentrazioni superiori dell’idrossido, la [Ca2+] in soluzione resta 0.011 moli/litro, mentre quella degli ioni OH– resta 0.022 moli/litro.
Il pH di Acidi e Basi deboli
Gli acidi deboli, in soluzione, sono parzialmente dissociati. Ovvero danno luogo ad un equilibrio come, ad esempio nel caso dell’acido acetico
![]()
L’equilibrio è molto spostato a sinistra. Per calcolare il pH di una soluzione di un acido debole è necessario conoscere quanto l’acido è dissociato. Il parametro che fornisce questa indicazione è la sua Ka, ovvero la sua costante di dissociazione acida. La Ka è definita attraverso la costante di equilibrio della reazione di dissociazione.
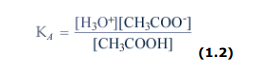
Vi ricordo che la concentrazione molare dell’acqua, che resta anche in questo caso costante, è contenuta nel valore di Ka, analogamente a quanto abbiamo sottolineato a proposito della Keq della dissociazione dell’acqua e di Kw.
Tanto maggiore è il valore della Ka, tanto maggiore è la tendenza dell’acido a dissociarsi, ovvero tanto più forte è l’acido. I valori di Ka degli acidi più comuni si trovano spesso tabulati sulle Tavole periodiche.
Prima di utilizzare l’equazione 1.2 per il calcolo del pH, è necessario fare un paio di considerazioni
-Poiché per ogni molecola di acido acetico dissociata vi è uno ione H3O+ e uno ione CH3COO–, queste due specie sono quantitativamente identiche: ovvero, [H3O+ ] = [CH3COO– ].
-Poiché l’acido è poco dissociato, la concentrazione dell’acido indissociato, che compare al denominatore nell’equazione all’equilibrio, può essere considerata uguale alla concentrazione analitica dell’acido, C°a. quindi, per un qualsiasi acido debole, la 1.2 diventa
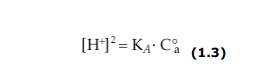
da cui
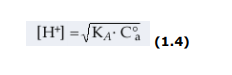
Basi deboli. Il caso è del tutto analogo a quello di un acido debole. L’ammoniaca è un classico esempio di base debole. Le proprietà basiche dell’ammoniaca non trovano giustificazione nella teoria di Arrhenius. In soluzione, l’ammoniaca dà luogo alla seguente reazione con l’acqua
![]()
Anche questo equilibrio è molto spostato a sinistra.
Le considerazioni fatte a proposito dell’acido acetico sono del tutto applicabili a questa reazione.
Ovviamente, in questo caso si parlerà di una costante di dissociazione basica e di ioni OH– invece che H+.
Infatti
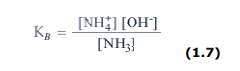
Da cui si ottiene che
![]()
in cui C°b indica in questo caso la concentrazione analitica della base.